
Everything about Polylactic Acid (PLA)
Indexes
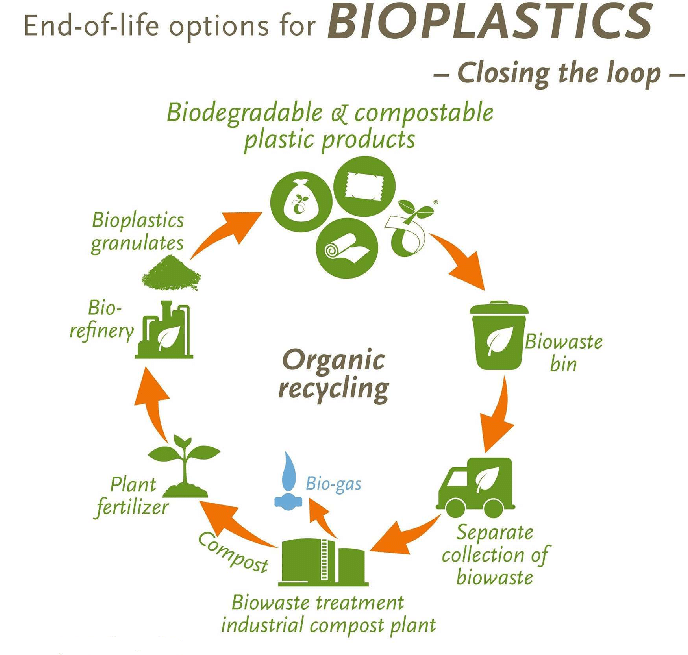
PLALEAVES products application
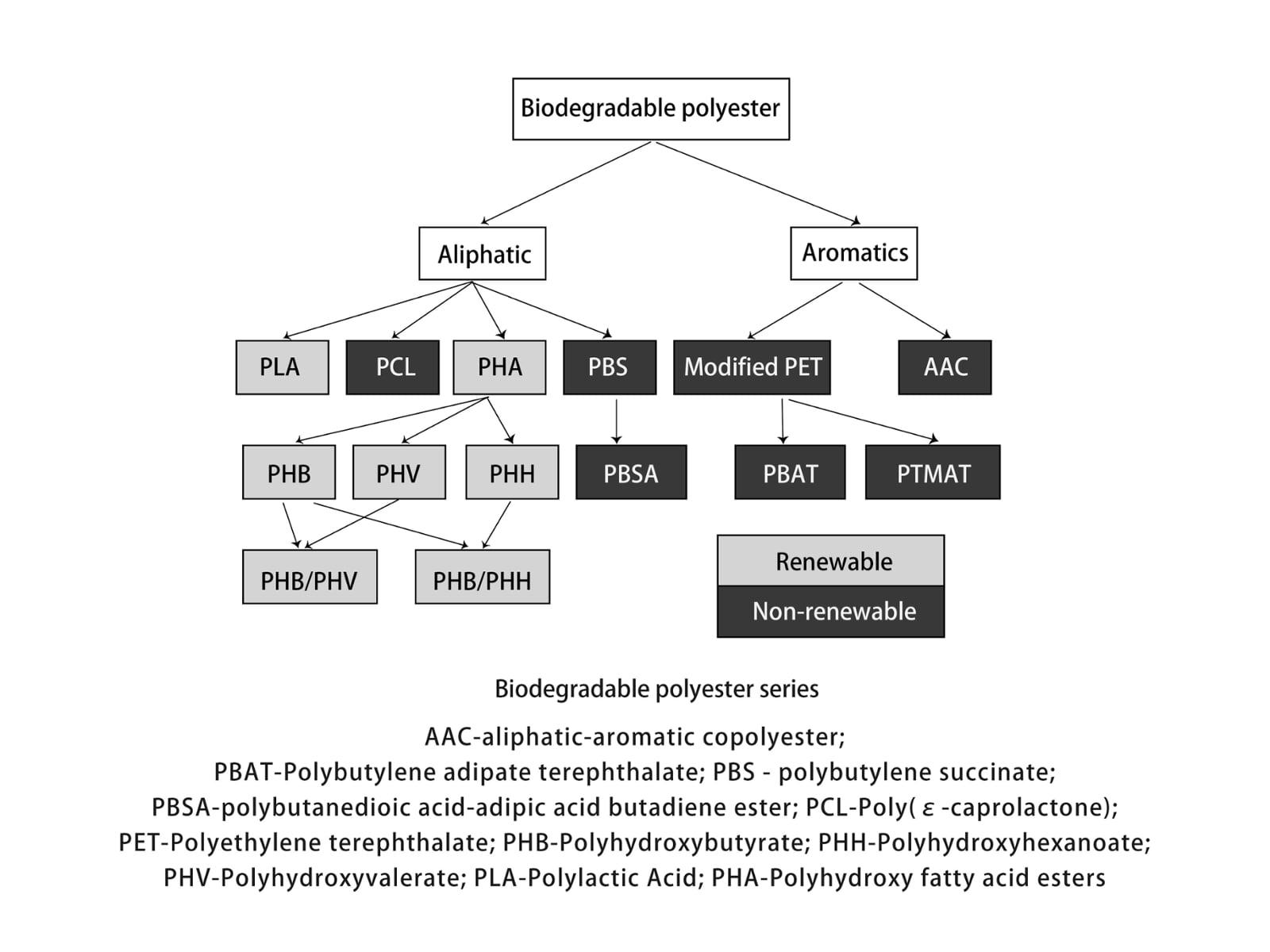
1. Overview of biodegradable polymers and Polylactic acid.
2. Synthesis and production of polylactic acid
3. Thermal properties of polylactic acid
4. Chemical properties of polylactic acid
5. Mechanical properties of polylactic acid
6. Rheological properties of polylactic acid
7. Degradation and stability of polylactic acid
8. Additives and processing aids for polylactic acid
10. Applications of Polylactic Acid
11. Environmental Assessment and International Standards for Polylactic Acid
7. PLA Bags
8. PLA Filament(For 3D Printers)
9. PLA Bottle
10. PLA Baby Tableware
11. PLA Food Packing Film
12. Medical Implants
1. Overview of biodegradable polymers and Polylactic acid.
1. Overview of biodegradable polymers and Polylactic acid.
1.1 Overview of biodegradable polymers.
One of the biggest problems of this century is environmental problem, which comes mostly from global warming and white pollution. In the recent decades, the volume of plastic waste has reached huge numbers and currently it poses serious problems for the Earth. To be able to cope with the growing number of plastics, many scientists and researchers started extensive studies in this field with a purpose to find ways to replace regular plastics in people’s lives and in different industries. One of the best solutions that has been studied deeply is Polylactic acid (PLA). PLA is an environmentally friendly and 100% bio based polyester; it is fully biodegradable and compostable. These properties make PLA a material that can perfectly replace fossil-based polymers.

Regular plastics have their benefits and advantages for their users compared to other materials, and that’s why people still use numerous kinds of plastic products in their daily lives. And despite of the fact that the plastic pollution is considered as one of the most serious problems for all the world, the global production volume of plastics was estimated around 359mln tons in 2018. Moreover, estimations show that until now the total production of global polymers has reached approximately 9bn tons. Unfortunately, numbers show that only a very small part from these plastic goods is recycled and reused. In particular:
· The share of polymers that have been recycled and reused is 9-10%
· The share of polymers that have been incinerated is 12%
· The share of polymers accumulated in oceans, lakes, rivers and landfills is 78-79%.
These facts show us that we face serious problems related to the disposal of regular plastics. The consequences of plastic waste accumulation include environmental pollution, high level of toxicity of ecosystem and increasing danger for human health.
The largest share from the global production of plastics belongs to packaging materials (35% – 45%). These include different types of Single Use Plastics (SUPs), for example packaging films, bottles, bags, cutlery, cups and straws. The main problem with these kinds of products is that due to their lightness, their ability of being recycled is restricted. This fact causes environmental problems, and the negative effect on environment is becoming even more serious in the result of huge concentration of plastic waste, its widespread distribution and non – biodegradable features. Researches show that in many cases, plastic goods do not completely degrade. Instead, they end up in oceans (approximately 13Mts in a year), where they are deteriorated to micro pieces (with diameters less than 5mm). These micro pieces pose a big danger for fish and other marine animals as they can be ingested by this wildlife and cause serious problems in food chain. Besides, the micro pieces can pass into human organism via drinking water. The impact of these micro pieces on human health is still unknown.
According to scientists that work in the polymers industry, this field has two major problems, which pose significant risks to the environment. One of those problems is huge number of carbon dioxide emissions, the second one is high level of fossil resources’ consumptions.
As number of researchers have concluded that one of the most efficient solutions to cope with plastic pollution is to use renewable resources to produce polymers, i.e. to produce biobased polymers, in recent years the global interest on biobased polymers has increased significantly. Despite of the fact that the demand for PET and other regular plastic is growing, the market of bioplastics is also a fast growing and developing sector. There is a list of companies and manufacturers, which conduct activities in the industry of biobased polymers. Their aim is to create new types of bioplastics that not only do not pose risks for the environment, but also are useful for the Earth. PLA, PGA with their copolymers, PHAs, PHB, PHV and their copolymers, etc. are currently the most investigated biodegradable polymers. The latest market research implemented by European Bioplastics in the partnership with the nova- Institute shows that global production of bioplastics is predicted to increase to around 2.87mln tons in 2025. To compare, in 2020 bioplastics production was estimated approximately 2.11mln tons, the 60% or over 1.2 tons out of which is biodegradable plastic. This number is achieved mainly due to the growing production of PLA and PHA. In particular, from 2013 to 2020, the production of PHA has increased from 5.3mln tons to 17.0mln tons. The global production of PLA was estimated 190,000 tons in 2019. PLA has a high potential for growth, as the annual volumes of investments in the production of this biodegradable polymer are growing, especially in US and Europe.
Taking into account the above – mentioned advantages of biodegradable polymers, scientists think that these polymers are an extremely important discovery and the use of biodegradable polymers will significantly contribute to the environmental protection. Biodegradable polymers are considered as innovative solutions and currently they are used in many industries starting from automotive and agriculture to packaging industries and biomedicine.
Currently, one of the best biodegradable polymers is Polylactic acid. Polylactic acid (PLA) due to its properties:
1. Biocompatible
2. Biodegradable
3. Bio-based
It is called the "polymer of the 21st century". One of the most important advantages of PLA is that it can be efficiently used in all the above-mentioned industries. First scientist who synthesized PLA with low molecular weight was Carothers (1932). Two decades later, another scientist, DuPont successfully made PLA with a higher molecular weight. PLA is made from lactic acid, which is animal or plant-based. The main sources of lactic acid are starch, corn, fish waste, etc. Currently, PLA is considered as the most eco friendly polymer and acts as a perfect alternative to the traditional petroleum-based polymers. The list of PLA applications are growing and creating more demand for its production.
PLA production process consists of the following steps:
1. Fermentation
2. Separation
3. Polymerization.
The monomers that are used to polymerize PLA, are taken from the nature. The main sources for these monomers are sugarcane, cornstarch and other renewable resources. Moreover, according to a US based company NatureWorks, currently it is also possible to reduce the costs of PLA production. PLA basically is being synthesized through the ROP (ring opening polymerization) of lactide monomer. There are different types of catalysts (for example, zinc, lead, aluminum, etc.) and solvents (for example, diphenyl ether, chloroform, etc.) that can be used during this process. In this way, it is possible to control the molecular weights of PLA polymer, as well asthe sequence and ratio of L- and D-lactic acid units in the final PLA polymer.
Polylactic acid has a list of advantages compared to other biobased polymers. PLA can be biodegraded into H2O and CO2, and both of these elements are non-carcinogenic and non-toxic. Due to these features, PLA has a high level of safety for human organism. High security level allows PLA to be used in different serious industries such as biomedicine. In this field, PLA polymer is used to make clips, sutures, drug delivery systems and so on. Another examples of industries that use PLA polymer are construction, agriculture, automotive, food packaging and so on.
Another advantage of PLA is that it has a good thermal processability. This property allows it to be configured by other technologies such as melt extrusion, cast film, blow molding and fiber spinning.
And the last but not least advantages is that PLA is a promising biodegradable polymer that can be used to develop such innovative solutions that require to use PLA composites and advanced manufacturing methods.
1.2 Market potential for biodegradable polymers and polylactic acid
PLA was discovered in the early 19th century when Pelouze concentrated lactic acid to form low molecular weight PLA through a process of distillation of water. This is an early lactic acid polycondensation process for the preparation of low molecular weight PLA and propylene-based esters. Propylene glycol is a prepolymer or intermediate used to convert high molecular weight PLA. This polycondensation process can produce small amounts of low purity PLA. Nearly a century later, Dupont scientist Wallace Caroth-ers discovered that PLA could be produced by heating propylene glycol in a vacuum. In addition, for high purity PLA, this process is not feasible on an industrial scale due to the high cost of purification, thus limiting the production of medical grade products such as sutures, implants and drug carriers. The ambitious Cargill Corporation has been involved in the development of PLA production technology since 1987, and first established a pilot plant in 1992. In late 1997, Cargill and Dow Chemical formed a joint venture called Cargill Dow Polymers, LLC to further commercialize PLA. Their efforts paid off with the launch of the Ingeo brand of products. As part of the joint venture, Cargill worked to improve the hardening time of PLA products, while Dow Chemical focused on the production of PLA. In general, monomeric lactic acid of PLA can be obtained by bacterial fermentation of glucose; and glucose is extracted from plant starch. Thus, PLA is a polymer made from renewable resources that has the potential to reduce our dependence on traditional plastics made from petroleum-based resources. In recent years, there has been tremendous progress in PLA research, with many inventions and products worldwide.
Plastics manufacturing is a major global industry. Every year, billions of tons of raw and recycled plastics are produced.
Polymer production has been increasing year by year except for 2008-2009 when plastic production decreased due to the global financial crisis. With the recovery of the world economy, the demand for plastics has quickly recovered. The world's largest plastics producers, Dow Chemical, ExxonMobil Chemical and BASF, are all proving to have double-digit growth in plastics sales and production. Dow Chemical reported a 15 percent increase in sales for all regions in the fourth quarter of 2010. This is due to the rapid growth of the automotive industry and the demand for elastomeric materials due to the growth in global automotive demand. BASF reported a 26 percent increase in sales in the first quarter of 2010, thanks to substantial growth in the automotive and electrical/electronic industries. In the first quarter of 2010, large chemical company ExxonMobil's sales rose 38% to $6.3 billion, and a large part of that came from the plastics business.
Overall, in 2009, a researcher estimated the global quantity demand to be 45 kg per capita by 2015. The plastics market is still a shared "pie" among existing producers, and there will be opportunities for latecomers to gain market share. According to research data provided by Accenture, a global management consulting firm. The highest growth area for polymers is in the electrical and electronics industry, where highly complex electrical and electronic products such as smartphones, computers and entertainment devices require durable and lightweight components, which makes polymers play a critical role in their design. Liquid and solid plastic products, including packaging,
toy containers and stationery, continue to be the most consumed industry for polymers, estimated at 78,361,000 tons per year.
These figures are strong evidence that the demand for plastic products will grow further in the future. However, the majority of polymers on the market are petroleum-based products. Since oil prices rose to $147 per barrel in July 2008, the prices of many oil commodities, particularly polymers, have reached historic highs, although crude oil prices have now recovered to affordable levels. Today, many believe that another oil price increase is likely in the next 10 years due to limited crude oil reserves. The continued exploitation of these natural resources is also contributing to severe global warming. Therefore, the search for alternative energy sources and non-oil products is essential for a sustainable economy and the environment.
As mentioned earlier, biodegradable polymers can be obtained from petroleum and renewable resources. Both types of biodegradable polymers have attracted the attention of industry. Petroleum-based biodegradable polymers help overcome the accumulation of non-degradable plastic waste. However, renewable biodegradable polymers are not only biodegradable but can also be obtained from sustainable resources with environmental credentials.
For environmental reasons, many countries have implemented regulations to reduce or ban the use of non-degradable plastics. For example, China, the largest consumer of polymers with a population of 1.4 billion, has restricted the use of plastic bags. Major supermarkets do not provide free plastic bags to customers.
These actions save at least 37 million barrels of oil per year. In Europe, a number of regulations promote organic waste management to help reduce emissions of toxic gases and greenhouse gases from soil/water. Recycling of biological waste is the first measure to reduce the production of methane (a greenhouse gas) from landfills. Decree 1999/21/EC on landfills requires EU member states to reduce the amount of biodegradable waste to 35 percent of 1995 levels by 2016. The second measure is to increase the use of compostable organic materials, which play a role in helping to enrich the soil. This helps to replace the carbon loss in soil highlighted in the Waste Directive 2008/98/EC (Waste Framework Directive). Following the requirements of Directive 94/62/EC on packaging and packaging waste, plastics and packaging waste should now comply with European Standard EN13432 and these materials should be declared compostable prior to marketing.
Ireland was one of the first countries to introduce a plastic bag tax. Ireland's Department of Environment, Heritage and Local Government imposed a 15-cent fee on plastic bags in 2002. This had an immediate impact, reducing the number of plastic bags used per capita from 328 to 21. Following this encouraging result, the Irish government raised the tax to 22 cents, further reducing the use of plastic bags. Although biodegradable plastic bags degrade faster than standard plastic bags, the Irish government does not distinguish between the two types of plastic bags in its laws. However, reusable plastic bags sold in stores are exempt from taxation, provided they are sold for no less than 70 cents.
Since the use of plastic bags cannot be completely avoided in modern life, it is recommended to use compostable materials to produce reusable plastic bags so that the disposal of waste will not be a burden on the environment. As awareness of compostable packaging grows, many companies are trying to make their products appear to have at least that packaging. As a result, there are various types of "eco-packaging" on the market. This eco-plastic product needs to undergo a series of tests to verify its biodegradability and compostability. In EU countries. Compostable packaging must meet the requirements of EN 13432, while other countries also have their own standards. Because of its biodegradability, PLA's application as a packaging material was initially focused on high-value food and beverage containers as well as cups, rigid thermoplastic foam, and high-cost films. Although PLA is a biodegradable polymer material, it can be used as a substitute for non-biodegradable polymers in packaging applications. However, its application as a packaging material is still very limited due to its high production cost.
In the last few decades. There has been a tremendous growth in the production of biodegradable polymers. Shen et al. (2009) conducted an overview of the products and markets for bio-based plastics (PRO - BIP 2009), and the global production of bio-based plastics was 360,000 tons in 2007, which represents only 0.3% of the total global plastics production. However, the production of bio-based plastics is growing rapidly, with an annual growth rate of 38% during 2003-2007 ( Shen et al. (2009). Shen et al. (2009) predicted that the production of bio-based plastics will increase to 3.45 million tons by 2020, mainly consisting of starch-based plastics (1.3 million tons), PLA (0.8 million tons), bio-based polyethylene (0.6 million tons) and PHA (0.4 million tons). Bio-based polyethylene is prepared from ethylene by dehydration of bioethanol produced by sugar fermentation process. A large number of bio-based projects have been initiated in the United States, Europe, Japan and other countries, and production has been transferred to other parts of the world.
According to European Bioplastic (2017), cellulose-based polymers account for the largest share of biodegradable polymers worldwide. Cellulose polymers are mainly used in the manufacture of fibers for textiles, bedding, mats, filter membranes, etc. Much of this cellulose comes from cotton and is chemically treated or modified to suit the end use. Starch-based polymers involve starch polymer blends and thermoplastic starches. Companies such as Novamont, Plantic DuPont and Cereplast blend starch with other synthetic polymers to improve the processing and mechanical properties of starch. Usually, the mixing of starch with biodegradable polymers (e.g. PCL, PLA or PHB) is more appropriate to ensure that the resulting mixture is fully biodegradable. Some manufacturers of starch-based polymers also blend starch with polyolefins, and these starch-polymer blends can partially degrade the starch and thus initiate degradation. However, the residual polymer backbone can still have harmful effects on the environment.
PLA is the most widely produced renewable biodegradable polymer. Currently, most renewable biodegradable polymers are still in the development stage. Due to the maturity of mass production technology, PLA has a large share in the market. Technologists also prefer PLA because of the carbon credits it offers for renewable raw materials. Well-known manufacturers, particularly NatureWorks, have established downstream processes and markets that are also facilitating the expansion of PLA production in a range of countries. It is expected that the production of PLA may exceed that of other biodegradable polymers combined, such as PBS, PBT, PCL and PBAT. Future mass production and market competition will also help develop economically viable technologies to deliver cheaper products. Investors may prefer high-volume production of PLA because of the known profitability of PLA and its long-term origin from low-cost agricultural raw materials. In addition, the development of starch-based and other bioplastics will also increase the demand for PLA. This is because fully biodegradable starch blended with PLA helps to improve the weak structural properties of the starch itself. Similarly, BASF's AAC Ecovio is an end product with better processing performance and flexibility when blended with PLA.
Although PLA was first synthesized in the early 19th century, it took a long time for the development of PLA to reach production feasibility. In the early stages of commercialization, PLA was limited to applications in biomedical devices due to the high cost of synthesis and the fact that it was not mass-produced. In order to obtain high molecular weight PLA, direct polycondensation needs to be controlled by a key process. In the 1990s, the market for PLA began to expand, and Cargill built its first pilot plant in 1992 to increase the yield of PLA using indirect polymerization of propylene-based monomers. In 1997, Cargill and Dow Chemical formed a joint venture to create Cargill Dow, Inc. and launched the first commercial products under the brand name Ingeo. In 2002, a plant was built in Blair, USA, at a cost of $300 million. Later, in 2007, Dow Chemical sold its 50% stake in NatureWorks to Teijin Corporation of Japan. Teijin has been developing green plastic technologies to expand its existing range of polymer resins. During the recession, Teijin restructured and transferred its 50% ownership to Cargill, and is now working on their PLA product BIOFRONT - a heat resistant PLA plastic that can replace PET. The melting temperature of BIOFRONT is 40°C higher than that of existing PLA. Teijin's BIOFRONT was produced in collaboration with Mazda to develop a car seat fabric made of 100% biofiber. Teijin and Panasonic Electric Works collaborated on the development of a PLA compound, MBA900H, with excellent malleability. Since Teijin's withdrawal, NatureWorks has become a wholly owned subsidiary of Cargill Corporation. In a March 2009 company press release, NatureWorks announced that the company was evaluating plans for a new facility to manufacture Ingeo. Ingeo is used by hundreds of well-known brands at retailers in the United States, Europe and Asia.

Corbion, formerly known as Purac, is currently the world's largest producer of lactic acid and still operates a lactic acid plant in Thailand with an annual production capacity of 120,000 tons in 2016. The entire plant plans to increase its annual capacity to 205,000 tons in the future. Corbion currently supplies more than 60% of the world's lactic acid from its plants in the Netherlands, Spain, Brazil and the United States. Corbion - Purac's PLA and PLA copolymers are used in biomedical applications such as sutures, staples, medical staples and tissue scaffold materials. As the PLA market matured, Corbion decided to use its lactic acid production for PLA production in order to further expand its business. The volume of lactic acid produced was large enough to allow Corbion to convert the lactic acid into L-propyleneglycerides and D-propyleneglycerides under the brand name PURALACT. Corbion invested 45 million euros in the production of 75,000 tons of PLA at its propyleneglyceride plant in Thailand.
Corbion of the Netherlands and Sulzer Chemtech of Switzerland have joined forces to produce PLA foams. Synbra is a company in Etten-Leur, the Netherlands, dedicated to solving the PLA foam technology for Purac-Sulze and expanding its product range to include BioFoam, a green polymer foam. synbra has been in the StyroFoam manufacturing business for over 70 years. Synbra's foamable PLA utilizes propylene glycol produced at Purac's propylene glycol plant in Spain, which will have a capacity of 1 x 107lb (1lb = 0.45kg) per year once it is fully operational. In September 2010, Purac partnered with Arkema to develop a high purity functional block copolymer containing PLA linkages using Purac's propylene-based esters. The result of this development is an improvement in the current propylene-based ester polymerization process, which is free of metal residues, thus ensuring the safety of packaging for medical consumer products. In addition, Purac has partnered with Toyobo, a Japanese film, fiber and biotechnology company, to produce an amorphous and biodegradable PLA product for the European market under the brand name Vyloecol. Vyloecol is a patented amorphous PLA used as a coating or binder for packaging films and materials.
Sulzer is also actively involved in PLA production in the EU with Galaxy Galactic and Total Petrochemicals. They formed a 50/50 joint venture, Futerro, in September 2007 to develop PLA technology. The initial project is the construction of a 15 million ton PLA capacity demonstration plant, a pilot unit costing $15 million. The Galaxy production site is located in Escanaffles, Belgium. Propyl cross ester monomer is extracted from sugar beet by fermentation. Another joint venture, Pyramid Bioplastics Guben GmbH, is also building and operating a plant for the production of PLA in Guben, eastern Germany. The company is a joint venture between Pyramid Technologies Ltd, Zug, Switzerland, and Guben Bioplastics GmbH, Germany. At that time, the first plant was expected to have an annual capacity of 60,000 tons by 2012. A pilot plant manufacturer, Hycail, was sold in 2006 to Tate & Lyle, which had produced a small amount of PLA; however, the plant was closed two years later.
In Asia, a number of companies have been established to explore PLA technology. Japan was the first country to participate in PLA research and development. As the market for PLA grew, China followed suit. Although Japan was involved in PLA technology earlier than other Asian countries, some ambitious large companies had to stop production due to high production costs, lack of raw materials, and an immature market that could not accept the high price of this high-quality plastic. Shizmadu initially operated a pilot plant to produce a small amount of PLA for commercial use, but the plant has also ceased production and the technology was sold to Toyota Motor Corporation. Toyota has increased its production to 1000t/ a, mainly for automotive applications. In 2008, the plant was sold to Teijin, and now Teijin is expanding production of BIOFRONT products. 2008, the company plans to increase BIOFRONT production capacity to 5,000t/ a in 2011. Unitika Ltd. is a 120-year-old textile company that markets PLA products under the Teramac brand name. Teramac resins can be molded using a variety of plastics processing technologies, including injection, extrusion, blow molding, foam molding and emulsion polymerization. Toray Korea has launched full commercialization of Ecodear PLA films and sheets. Ecodear offers heat resistance, impact resistance, flexibility and high transparency comparable to petroleum-based plastic films.
1.3 General properties and applications of polylactic acid
1.3.1 Household polylactic acid
Nature Works is the world's largest producer of PLA. Its product range includes injection molding, extrusion, blow molding, thermoforming, film and fiber applications. NatureWorks' PLA resin, Ingeo, is produced at a rate of 140,000 tons per year at its Nebraska, USA facility. The company has 19 sales locations worldwide to market and promote their products. NatureWorks has launched a co-branding program to better position Ingeo in the marketplace. Currently, more than 900 companies are participating in this partnership program, successfully strengthening Ingeo's global brand position.
The choice of Ingeo is determined by the process and the end use of the product. According to NatureWorks CTO Patrick Gruber and colleagues, the various grades of PLA are based on the principles of chemical structure purity, molecular weight and additive quantity. Controlling the chemically structured composition of PLA has a significant impact on the melting point, crystallization rate, and ultimate degree of crystallization. The melting point of PLA in pure L or D chemistry is 180°C and the glass transition temperature is 60°C. Copolymerization of D-propyl or racemic propyl esters affects the purity of the chemical structure. The crystallinity of PLA was completely destroyed by the addition of 15% of racemic or D-propyl cross-ester to poly(L-lactic acid) (PLLA). The copolymerization of L and D chemical building blocks results in a polymer with an amorphous structure. However, in order to avoid thermal deformation of the PLA used in hot food packaging containers, the higher the melting point of the resulting polymer, the better. Plack claims that by controlling the stereocomplex and steric block of propylene cross-ester during copolymerization, the melt temperature can be effectively increased to 230°C, almost as good as that of polystyrene (which has a melting point of about 240°C). Nevertheless, it is very important that the rheological properties of the resulting polymer are compatible with the processing.

PLA is a typical aliphatic polyester with relatively poor strength and lack of shear sensitivity. The introduction of branched chains into PLA can result in longer polymer chains and better entanglement, thereby improving melt strength during film blowing. However, the details of such modifications are rarely disclosed by manufacturers. Further details of the research work on the rheological properties of PLA are discussed in the context of PLA synthesis and production.
Unigyco Ltd. and FKuR Plastics Ltd. market their NatureWorks Ingeo-based products under the brand names BioFlex and Terramac, respectively. While both manufacturers emphasize that their products are based on Ingeo, some modifiers or additives have been added to the products to improve the original properties of the PLA. Improvements to PLA by other manufacturers are seen as positive steps toward making PLA available to a wide range of market needs. The Unigyco Terramac line also includes PLA foams and emulsions. The goal of PLA foams is to replace Stryrofoam, while reducing environmental pollution. Emulsion-grade PLA is suitable for coating agents. Similarly, Toyota produces PLA under the brand name Vyloecol, which is primarily used as a general purpose coating agent.
1.3.1 Biomedical polylactic acid and its copolymers
In addition to the production of environmentally friendly household products to replace existing petroleum-based plastics, PLA is also widely used in the biomedical field for the production of bioabsorbable implants and devices for internal fixation after fracture due to its biodegradability and good mechanical properties. In addition, it is also used as a controlled drug delivery vehicle in biomedicine due to its biocompatibility, biodegradability, non-toxicity and low immunogenicity. Most PLAs used in biomedical applications are produced from lactic acid. Phytochemicals made from poly-L-lactic acid are readily degraded and absorbed by the body by the action of enzymes. Unfortunately, the stereoisomer D-lactic acid is not degraded by enzymes in vivo. However, prolonged hydrolysis in body fluids will eventually break down most of the poly-D-lactic acid. This degradation mechanism will be discussed later in the synthesis and production.
A large number of PLA copolymers have been synthesized for tissue engineering. The main purpose of synthesizing these copolymers is to fine-tune the degradation cycle from a few weeks to several years. Usually, ethylene and e-caprolactone monomers are copolymerized with propanediol. The mass of poly-L-lactic acid was significantly increased after copolymerization with ethyl cross-ester and e-caprolactone. This has important implications for the preparation of tissue engineering scaffolds and trauma dressings. The copolymers are degraded to match the tissue growth and the loss of mass and strength of the prescribed implant. Eventually, the scaffold structure will be replaced by the patient's permanent tissue.
PLA and its copolymers are used in a variety of biomedical applications such as sutures, fixators, screws and stents, and they are used in oral, orthopedic, auricular and frontal plastic surgery. Screws and fixators are produced by injection molding. Sutures, produced by fiber spinning process. Bioresorbable scaffolds are fabricated using a range of techniques including phase separation, solvent evaporation, ablation casting/salt impregnation and fiber bonding to form polymer networks, PLA copolymers are also widely used as drug carriers and such drug carriers contain active drugs that are efficiently delivered to targeted cells and then released at controlled rates. Norelide is one of the best-known products on the market, a polymeric acid-co-ol ester containing goserelin as a controlled-release drug for the treatment of breast cancer. Norelide releases the drug slowly to inhibit the growth of hormone-dependent cancer cells. The U.S. Food and Drug Administration has also approved Norelide for the treatment of prostate cancer. There are other drug delivery systems on the market that are associated with PA tools. Purac is a major global manufacturer of biomedical and pharmaceutical release control grade PLA and its copolymers; its trade name is Purasorb. Durect also markets a bioresorbable polymer under the trade name Lactel. As can be seen from the properties of both manufacturers' grades, PLA copolymer is the most widely produced grade. All grades are tested for their intrinsic viscosity as a guide to the molecular weight of the polymer to be synthesized. This is very important in biomedical applications because it ensures the proper rate of absorption by the body. When the polymer is exposed to aqueous media or tissues, the ester bonds of the polymer react with absorbed water through a hydrolysis reaction. Over time, the long polymer chains are broken down into shorter chains, forming water-soluble fragments. Eventually, the water-soluble fragments diffuse from the initial polymer structure and are hydrolyzed into glycolic and lactic acids for hepatic metabolism. In general, the degradation rate is higher when the molecular weight is low and the ethylene cross ester content is high. Overall, the contribution of PLA and its copolymers to the medical industry has been significant.
1.4 Summary
PLA has been around for decades, but only in recent years has its application growth expanded rapidly. PLA is a biodegradable polymer with the potential to replace existing petroleum-based commodity polymers and help overcome the accumulation of plastic waste in landfills. In addition to its applications in general products and packaging products, it has biomedical applications in surgery due to its compatibility with living tissue. PLA is favored because it can be produced on a large scale from renewable agricultural resources, thereby reducing society's dependence on petrochemicals. Continued research and development has made it possible to produce few greenhouse gas emissions associated with the production process. In conclusion, PLA, as a biodegradable polymer, has great potential and market prospects for future sustainable development.
2. Synthesis and production of polylactic acid
2.1 Introduction
Polylactic acid (PLA) is synthesized from lactic acid (LA) monomers. Polylactic acid is produced by direct polycondensation (DP) and ring-opening polymerization (ROP). Although DP is simpler to produce than ROP, ROP can produce brittle PLA with low molecular weight. In general, the production of PLA involves several substances, which are related to each other as shown in Figure 2-1.LA in this process is obtained from the fermentation of sugar. la is converted to propanediol and eventually to PLA. It should be noted that there are two different terms for polymers of lactic acid, "poly (lactic acid)" and "polylactide" (also known as polypropylene glycol). The two terms are used interchangeably; however, there is a scientific difference because "polylactic" is produced via the ROP route, while "poly (lactide acid)" is produced via the DP route. In general, "poly (lactic acid)" is widely used to refer to polymers produced by LA [the difference between "poly (lactic acid)" and "polylac-tide" is explained here polylac-tide"].

2.2 Production of lactic acid
LA is the basic building block for the production of PLA. It is chemically known as 2-hydroxypropionic acid and has the chiral stereoisomers L (levorotatory) (-) and D (dextrorotatory) (+). The physical properties are listed in Table 2-1. Naturally occurring LA is mostly in the form of L, while chemically synthesized lactic acid can be a mixture of racemic D and L. LA is a biostable substance with high water solubility. Before LA was used in large quantities in biodegradable polymers, it was widely used in industry as a metal cleaning solvent, detergent, humectant, mordant and tanning agent. Its use as a humectant means that it acts as a moisturizer in cosmetics and personal hygiene products, while its use as a mordant is related to its use as an additive in the dyeing process to improve the acceptance of dyes in textiles. LA is also added in the manufacture of paints and inks for better adhesion to the printed surface. It is also used in the food industry to provide a sour taste to beverages. The addition of LA in the form of calcium lactate can extend the shelf life of meat, poultry and fish by controlling the growth of pathogenic bacteria while maintaining the original flavor of the food. Many dairy products, including yogurt and cheese, taste a bit sour, also due to the addition of LA, which provides additional antibacterial action in these products.
Table 2-1 Physical properties of lactic acid
Projects | Indicators | Projects | Indicators |
CAS Registration number | 50-21-5(D,L-Lactic acid) 79-33-4(L-Lactic acid) 10326-41-7(D-Lactic acid) | Taste Melting point/。C Boiling point/。C | Mild sour taste 53 >200 |
Chemical formula | C3H6O3 | Solubility in water /(g/100g H2O) | miscible |
Chemical name | 2-Hydroxypropionic acid | Solving for departure constants Ka | 1.38×10-4 |
Molecular weight | 90.08 | pKa | 3.86 |
Appearance | Aqueous solution | Ph(0.1%Solution,25。C) | 2.9 |
2.1 Introduction
Polylactic acid (PLA) is synthesized from lactic acid (LA) monomers. Polylactic acid is produced by direct polycondensation (DP) and ring-opening polymerization (ROP). Although DP is simpler to produce than ROP, ROP can produce brittle PLA with low molecular weight. In general, the production of PLA involves several substances, which are related to each other as shown in Figure 2-1.LA in this process is obtained from the fermentation of sugar. la is converted to propanediol and eventually to PLA. It should be noted that there are two different terms for polymers of lactic acid, "poly (lactic acid)" and "polylactide" (also known as polypropylene glycol). The two terms are used interchangeably; however, there is a scientific difference because "polylactic" is produced via the ROP route, while "poly (lactide acid)" is produced via the DP route. In general, "poly (lactic acid)" is widely used to refer to polymers produced by LA [the difference between "poly (lactic acid)" and "polylac-tide" is explained here polylac-tide"].
2.2 Production of lactic acid
LA is the basic building block for the production of PLA. It is chemically known as 2-hydroxypropionic acid and has the chiral stereoisomers L (levorotatory) (-) and D (dextrorotatory) (+). The physical properties are listed in Table 2-1. Naturally occurring LA is mostly in the form of L, while chemically synthesized lactic acid can be a mixture of racemic D and L. LA is a biostable substance with high water solubility. Before LA was used in large quantities in biodegradable polymers, it was widely used in industry as a metal cleaning solvent, detergent, humectant, mordant and tanning agent. Its use as a humectant means that it acts as a moisturizer in cosmetics and personal hygiene products, while its use as a mordant is related to its use as an additive in the dyeing process to improve the acceptance of dyes in textiles. LA is also added in the manufacture of paints and inks for better adhesion to the printed surface. It is also used in the food industry to provide a sour taste to beverages. The addition of LA in the form of calcium lactate can extend the shelf life of meat, poultry and fish by controlling the growth of pathogenic bacteria while maintaining the original flavor of the food. Many dairy products, including yogurt and cheese, taste a bit sour, also due to the addition of LA, which provides additional antibacterial action in these products.
Table 2-1 Physical properties of lactic acid
Projects | Indicators | Projects | Indicators |
CAS Registration number | 50-21-5(D,L-Lactic acid) 79-33-4(L-Lactic acid) 10326-41-7(D-Lactic acid) | Taste Melting point/。C Boiling point/。C | Mild sour taste 53 >200 |
Chemical formula | C3H6O3 | Solubility in water /(g/100g H2O) | miscible |
Chemical name | 2-Hydroxypropionic acid | Solving for departure constants Ka | 1.38×10-4 |
Molecular weight | 90.08 | pKa | 3.86 |
Appearance | Aqueous solution | Ph(0.1%Solution,25。C) | 2.9 |
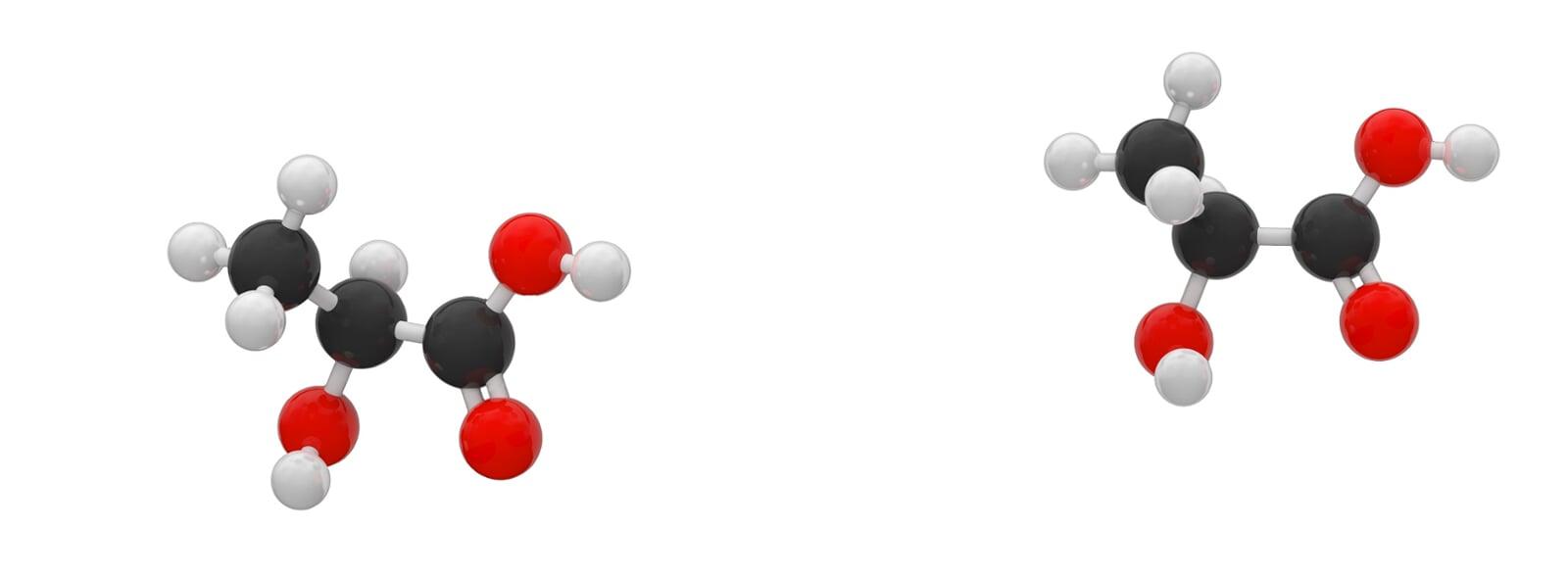
LA was first discovered by Carl Wilhelm Scheele in 1780, and the earliest technology was invented by the French scientist Frémy in 1881, since when it has been produced industrially using the fermentation process. Pure lactic acid has two stereoisomers (also called enantiomers), as shown in Figure 2-2. These two stereoisomers are synthesized by different lactate dehydrogenases in living organisms. Currently, 85% of LA is consumed by food-related industries, while the remainder is used in non-food applications, such as biopolymers, and solvent production.LA and lactate are naturally present in mammals when glycogen (a sugar stored in mammalian cells) is used anaerobically by muscles to produce energy (i.e., in the presence of insufficient oxygen supply). Although LA and lactate produced by muscles during anaerobic exercise can cause fatigue and pain, lactate has been found to be an important chemical used for sustained exercise. Lactate is produced by one muscle as fuel and can be easily consumed by another muscle. The soreness is due to the accumulation of acidic ions caused by the glycolytic reaction.
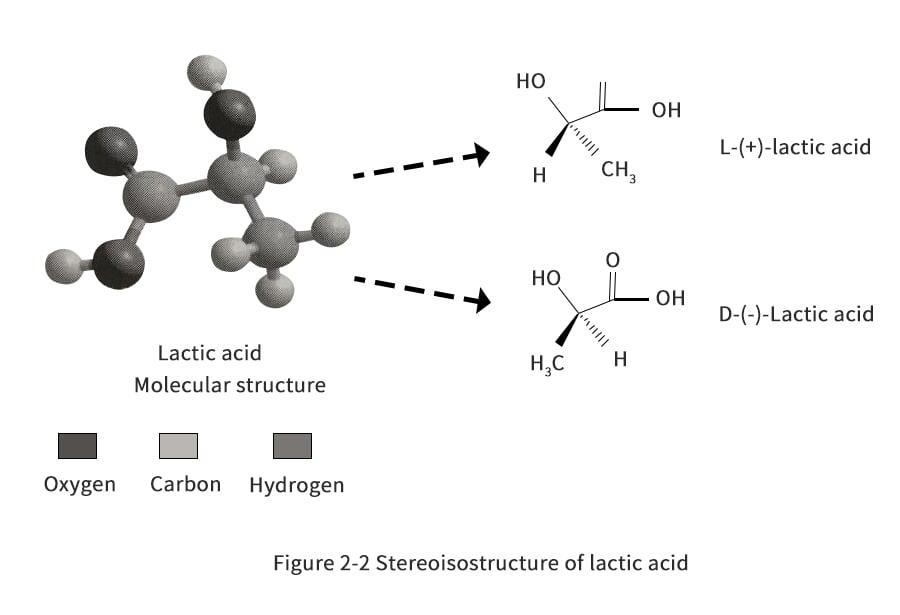
L-lactic acid can be metabolized in the body by enzymatic action. However, caution should be exercised when consuming D-lactic acid: according to the FAO/WHO guidelines, the maximum daily intake for adults is 100 mg/kg, and strictly speaking, infant foods should not contain D-lactic acid or D, L-lactic acid. Although the body does not produce D-lactase, small amounts are considered safe to ingest because the high solubility of D-lactate promotes hydrolysis in body fluids and subsequent excretion by the body's excretory system.
Most of the LA produced globally is made through a fermentation process. According to a review paper on LA development, there are about 20 LA-producing genera in the phylum Thick-walled Bacteria; these bacteria include Lactococcus, Lactobacillus, Streptococcus, Leucomonas, Soccer, Aerococcus, Carnotobacter, Enterococcus, Estrobacter, Tetraploidococcus, Vagalactococcus and Weiss. Among the many genera that produce LA, Lactobacillus is the most important, with about 80 species. These include Lactobacillus amylophilus, Lactobacillus pasteurii, Lactobacillus casei, Lactobacillus maltophilia, and Lactobacillus salivarius. The strains of Lactobacillus deuterium, Lactobacillus yanteri and Lactobacillus acidophilus produce both D-lactic acid and a mixture of the two stereoisomers.
While bacterial organisms and sugars are essential components of the fermentation process, organisms require a variety of nutrients to ensure their healthy function, including vitamin B, amino acids, peptides, minerals, fatty acids, nucleotides and sugars. The amount depends on the species and the source of these nutrients can be agricultural derivatives such as corn pulp and yeast extracts. LA bacteria are heterotrophic, which means they lack the ability to biosynthesize. The addition of complex nutrients significantly increases production costs, but the purity of lactic acid is higher.
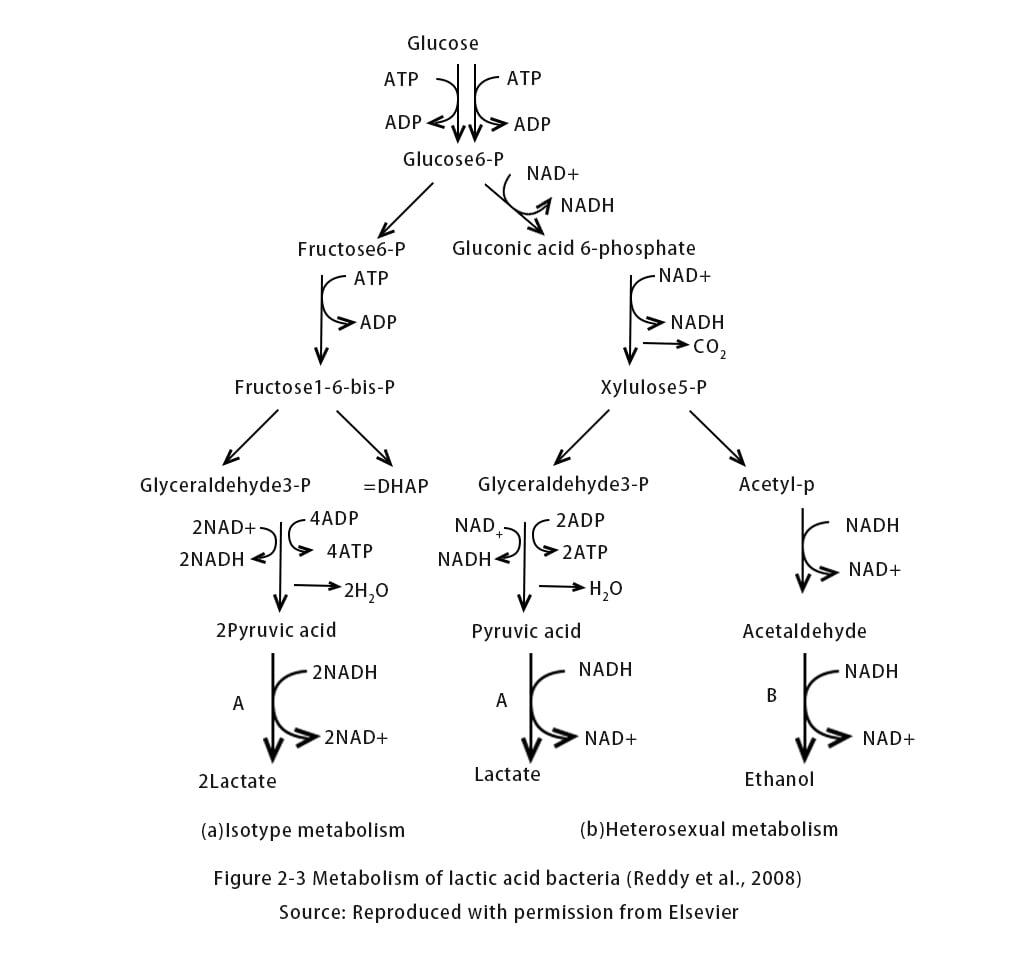
During LA fermentation, LA bacteria grow under anaerobic conditions in a low energy production mode. Compared to respiratory bacteria, this low capacity bacterium grows slowly. LA bacteria survive well at temperatures between 5 and 45°C and under mildly acidic conditions (pH=5.5-6.5). Reddy et al. classified Lactobacillus spp. into three groups based on their fermentation patterns. shows the products for each model. Fermentation of different types of sugar-rich substances altered the yield of LA (). In addition to lactic acid bacteria, there are other microbial sources of fungi such as Miscanthus mirabilis that can also produce lactic acid, but under aerobic conditions. However, the slow growth and low productivity of these fungi are detrimental to fermentation and require extensive agitation and aeration, resulting in high energy costs for long-term operation. Despite the focus on the use of wild microorganisms for LA production, attempts have also been made to increase L-lactate production through metabolic engineering.
Table 2-2 Lactic acid (LA) yields of starch and cellulose materials corresponding to microorganisms
Substrate | Microorganisms | LA Mass production |
Wheat and rice bran | Lactobacillus sp. | 129g/L |
Corn cob | Rhizoctonia sp. MK.96-1196 | 90g/L |
Pre-treated wood | Lactobacillus delta | 48~62g/L |
Cellulose | Lactobacillus rotundus ssp.Torquens | 0.89g/g |
Barley | Lactobacillus casei NRRLB-441 | 0.87~0.98g/g |
Cassava bagasse | German Lactobacillus NCIM 2025,Lactobacillus casei | 0.9~0.98g/g |
Wheat starch | Lactococcus lactis ssp. ATCC 19435 | 0.77~1g/g |
Whole wheat | Lactobacillus and Lactobacillus deuterium | 0.93~0.95g/g |
Potato starch | Miscanthus migrans, | 0.87~0.97g/g |
Corn, rice, wheat starch | Lactobacillus amyloliquefaciens ATCC 33620 | < 0.70g/g |
Corn Starch | Branched chain starch NRRL B-4542 | 0.935g/g |
The bulk commercial fermentation process for most LAs takes 3 to 6 days to complete; the sugar concentration used is between 5% and 10%. Many LA fermentation processes have been patented over the decades. Most of these patents keep the fermentation process confidential, but they also provide LA separation techniques. In US patent 6319382 B1, inventor Norddahl added whey protein as a nutritional substrate for LA bacteria and added protease to the fermenter to hydrolyze the protein during fermentation and thereby supply amino acids. In addition, the aqueous media used included yeast extract, K2HPO4, MgSO47H2O, MnSO4-2H2O, Tween80, lactose and cysteine hydrochloride to ensure optimal reactivity of the lactic acid bacteria. During fermentation, the pH of the aqueous slurry is monitored to maintain mildly acidic conditions close to neutral. The aim is to avoid the accumulation of LA in the fermentation medium and thus to inhibit the production of bacteria. Therefore, the successive addition of bases such as calcium hydroxide, sodium hydroxide or ammonia helps to convert the generated LA to lactate. The lactate can then be converted to LA by reaction with acid. Ammonia is preferable to other bases because it has the advantage of providing nitrogen nutrients to bacteria. This indicates faster bacterial growth compared to sodium hydroxide. Most processes use calcium hydroxide to control the pH of aqueous mixtures, including those used by NatureWorks. Sulfuric acid is then added to the LA medium to recover the LA, resulting in the formation and precipitation of gypsum (i.e., CaSO4.2H2O). The gypsum is separated from the medium by filtration and this gypsum is a by-product that can be sold as a building material or soil amendment. It is estimated that for every 1 ton of Los Angeles produced, 1 ton of gypsum can be produced.
The LA fermentation broth in the fermenter needs to be thoroughly separated to recover pure LA. Separation methods include electrodialysis, reverse osmosis, liquid extraction, ion exchange acidification, ion exchange purification, distillation, insoluble salt process or esterification. A comprehensive summary of LA purification techniques and their advantages and disadvantages is given by Henton et al. (Table 2-3). Although there is no difference between recovering D-lactate and L-lactate, extreme conditions (e.g. high temperatures) should be avoided because of the high potential for interconversion of D-lactate and L-lactate, resulting in the formation of racemic mixtures. To meet stringent oral requirements, food and pharmaceutical applications require highly optically pure L-lactic acid (>99%). Because LAs with different spinophobicity have different effects on the melting point, mechanical strength and degradation properties of PLA, single optical LAs are preferred for quality control.
Table 2-3 Lactic acid purification technology
Technology | Features | Advantages / Disadvantages |
Electrodialysis
| Can be used for continuous removal of lactic acid by a potential-driven membrane (LA) (lactic acid ion) | ①No acid fermentation required; ②Energy costs and funding |
Reverse Osmosis
|
Continuous LA removal by membrane
| ①Higher production due to the ability to maintain low acid levels in the fermenter higher rates; ②Membrane scaling; ③Needs acidic pH stable organisms |
Liquid Extraction
| from fermentation broth or acidified broth by preferential distribution to solvents Continuous removal of LA from
| ①Suitable for continuous processes and can effectively remove many non-acidic gases; ②High cost of capital; ③Cost of solvent loss |
Ion exchange (acidification) | Acidification of lactate by strong acid ion exchange resin
| ①Eliminates the need to add strong acids to the fermentation; ②Resin cost and resin regeneration issues |
Ion exchange (purification)
| LA is removed from aqueous solutions by complexation with amino-containing resins Removal from aqueous solution
| ①This is equivalent to the extraction technique of solid tertiary amines without solubility loss of; ②Resin regeneration; ③Cost and availability of resins |
Distillation method
| Separation of LA from volatile components by vacuum steam distillation
| ①LA can be distilled with water vapor; ②Must be heavily purified prior to distillation; ③Depending on the conditions, some degradation and oligomerization may occur |
Insoluble salt process
| The concentration of the fermentation or purification process should exceed the separation and acidification solubility of the lactate (e.g. CaSO4) | ①A simple process for utilizing low-cost capital; ②The crystallization of CaSO4 can entrap magazines and produce relatively impure acid |
Esterification
| Preparation of lactic acid esters and distillation of volatile esters
| ①Distillation and separation of esters to obtain high quality products; ②Need to convert acid |
Currently, NatureWorks has the largest single LA production facility, producing 180,000 tons of LA per year from corn. LA produced by NatureWorks is primarily used for conversion to Ingeo PLA. At the same time, Placid is the largest producer of LA for a wide range of applications in the food, beverage and pharmaceutical industries, as well as PLA mainly for surgical applications such as sutures, staples and screws. Placid has also been involved in studies on the copolymerization of LA with other monomers, such as ethyl cross-ester, ε-caprolactone or DL-lactic acid. The company has built a new LA plant in Thailand, which has been in operation since 2007. Using locally harvested sugar cane as raw material, the plant's initial capacity is designed to be 100,000 tons. Although LA is mainly produced using inexpensive agricultural raw materials, two companies still use chemical synthesis to produce racemic mixtures of LA: Musashino in Japan and Sterling Chemical Company in the United States. Chemical synthesis and common fermentation processes undergo different reaction pathways. An overview is given below.
Chemical synthesis methods:
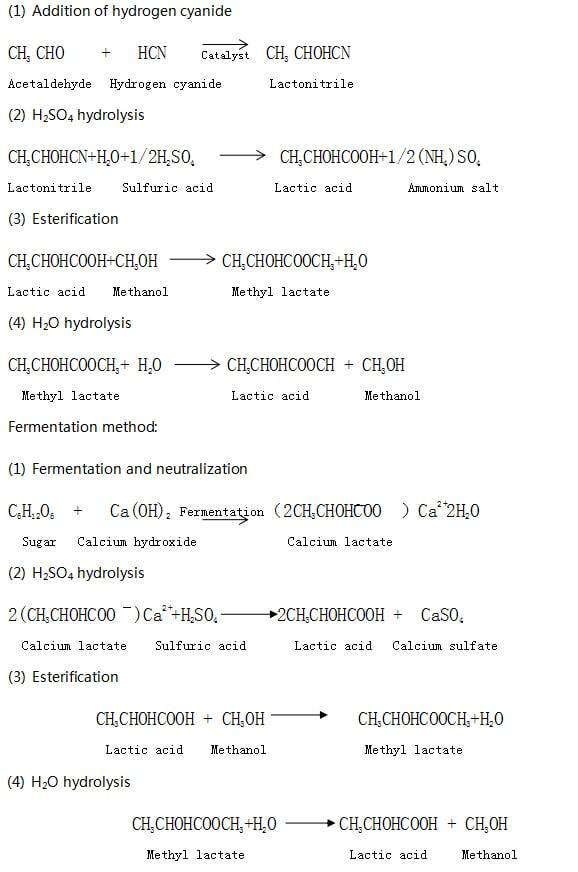
Commercially purified lactic acid is sold at a concentration of 50% to 80%. Typical food grade LA concentrations vary depending on the carbohydrate with which they are mixed, primarily to improve taste, nutritional value, or as a preservative. Food grade lactic acid produced by Galactic S.A., one of the major manufacturers of LA in Europe.Industrial grade LA for small-scale applications is sold in aqueous solutions at 80% to 88% purity. Examples include phenolic resin terminators, alkyd resin modifiers, solder fluxes, lithographic and textile printing developers, adhesives Youji, electroplating and electropolishing baths or detergent aids.
Pharmaceutical grade LA sells for $1000 to $1500 per ton, while industrial grade LA can be 20% cheaper depending on the application. Due to the maturity of implementing efficient fermentation processes and the demand for PLA in the local market, many new LA producers in China have not yet proven their viability in the short term. However, from a long-term perspective, the multiple uses of LA will maintain its market interest. Fermentation is the most common method used for the production of LA. The method outlined here can be used to synthesize LA in the form of lactic acid for the production of prepolymer LA.
The synthesis of lactic acid in the form of propyl cross ester is as follows:
(1) First, prepare 5 L of medium containing 500 g of glucose, 100 g of yeast extract and 100 g of polypeptone. The medium was sterilized using an autoclave and finally implanted with microorganisms from one of the following mobile genera: Lactobacillus, Streptococcus, Rhizobium, Bacillus or Leucomycetes.
(2) The mixture was incubated at 37°C and the pH was maintained at 7.0 using 6mo1/L ammonia. 15h was required to complete the incubation.
(3) The cultures were concentrated using 1000 g of ethanol and refluxed in a condenser between 90 and 100 °C for 3 h to obtain ethyl lactate.
(4) The consumed ammonia is separated with a gas scrubber bottle attached to the end of the condenser and cooled with ice water. The ammonia capture system is capable of collecting up to 98% of the ammonia gas.
(5) The remaining reaction mixture was kept at 80 °C to evaporate 750 g of unreacted ethanol by distillation.
(6) The reaction mixture was further raised to a temperature of 120 °C to remove water.
(7) After removal of water, the reaction mixture was distilled at a pressure of 50 mmHg and a liquid temperature of 70-100 °C to obtain 650 g of purified ethyl lactate for the condensation process (1 mmHg = 0.1333 kPa).
As shown in step (3), ethanol reacts with fermented LA through esterification to produce ethyl lactate (commonly referred to as lactate). The reason why lactic acid esters are more suitable than LA for conversion to lactic acid prepolymers is that LA is corrosive.
Therefore, the synthesis of PLA from lactate can help reduce costs by avoiding investments in corrosion-resistant reactors and equipment. In the long run, this means a significant cost reduction.
2.3 Production of propylene glycol ester and polylactic acid
Propylene glycol is an intermediate substance for the production of PLA by the ROP method. As can be seen in Figures 2-4, although both DP and ROP involve the production step of LA prepolymer, polymerization can be accomplished by forming propyl cross-esters without the use of coupling agents. The role of coupling agent is to increase the molecular weight of PLA, in fact, LA prepolymer is low molecular weight PLA (Mw= 1000 to 5000). This low molecular weight PLA is not usable, it is low strength, glassy and fragile. According to Garlotta, the direct reaction of prepolymers mainly uses low molecular weight PLA because of the lack of reactivity of the end groups once the polymerization is finished, the excess of water and the high viscosity of the polymer melt. Carothers first performed ring-opening polymerization of propylene glycol esters in the mid-20th century, and DuPont later revived the patent for this technology and began mass production of PLA. Depending on the type of initiator selected, propyleneglyceride molecules can undergo anionic or cationic ring-opening polymerization. The formation of free radicals in the presence of initiators promotes the chain reaction, resulting in the formation of high molecular weight polymers.
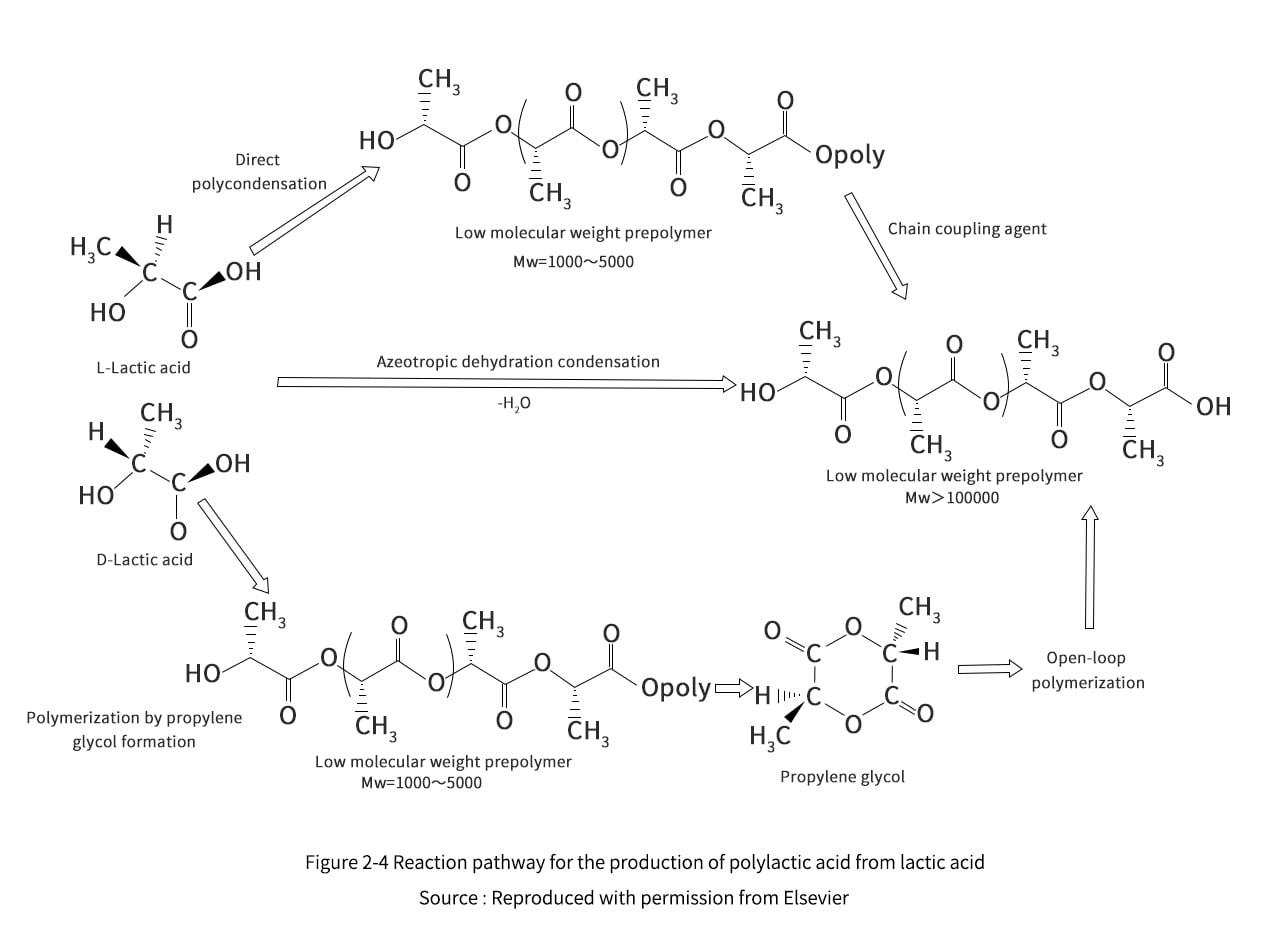
2.3 1 Prohexadione Production Technology Review
Since the 1930s, the technology for the production of propylene glycol esters has been studied, and Carothers et al. (1932) published a paper on the reversible polymerization of six-membered cyclic esters. Then, propyleneglyceride technology went through a period of inactivity because the purity of propyleneglyceride was not sufficient for mass production. The propylene glycol ester technology developed well after DuPont developed a purification technique. This eventually led to the mass production of NatureWorks. This section focuses on the early large-scale production of propyleneglycerides at Cargill-DuPont (now known as NatureWorks), and some related propyleneglyceride technologies.
U. S. Patent 5274073, entitled "Continuous Process for the Production of Pure Propyl Ester," invented by Gruber et al. (1993), describes a method for the production of propyl ester that can be summarized in the steps shown in Figures 2-5. Initially, the crude LA is fed to the evaporator. Typically, this is commercially produced LA, consisting of 15% LA and 85% water. This solution is made because the fermentation process is carried out in an aqueous medium. The evaporator is used to evaporate the water as top product, while the rest is concentrated LA. The LA produced by fermentation contains other impurities mixed with the enantiomers of L- and D-lactic acid. These impurities include sugars, proteins, amino acids, salts, metal ions, aldehydes, ketones, carboxylic acids and carboxylic acid esters, which may affect the quality of the production of propyleneglycerides and, consequently, PLA. Therefore, an evaporator can be designed to meet its purity requirements according to the specific practical situation. However, conventional evaporators, such as multi-effect evaporators, scrapers or falling film evaporators, can provide a basic separation of crude LA. It is preferable to operate the evaporator at less than atmospheric pressure to reduce energy consumption during heating, and it is also important to avoid racemic steric complexes of D-propyl cross-ester, L-propyl cross-ester or racemic propyl cross-ester (Figure 2-6), which tend to cause quality problems during polymerization to form poly(D,L-lactic acid). By the time it leaves the evaporator, the crude LA is concentrated to more than 85%.
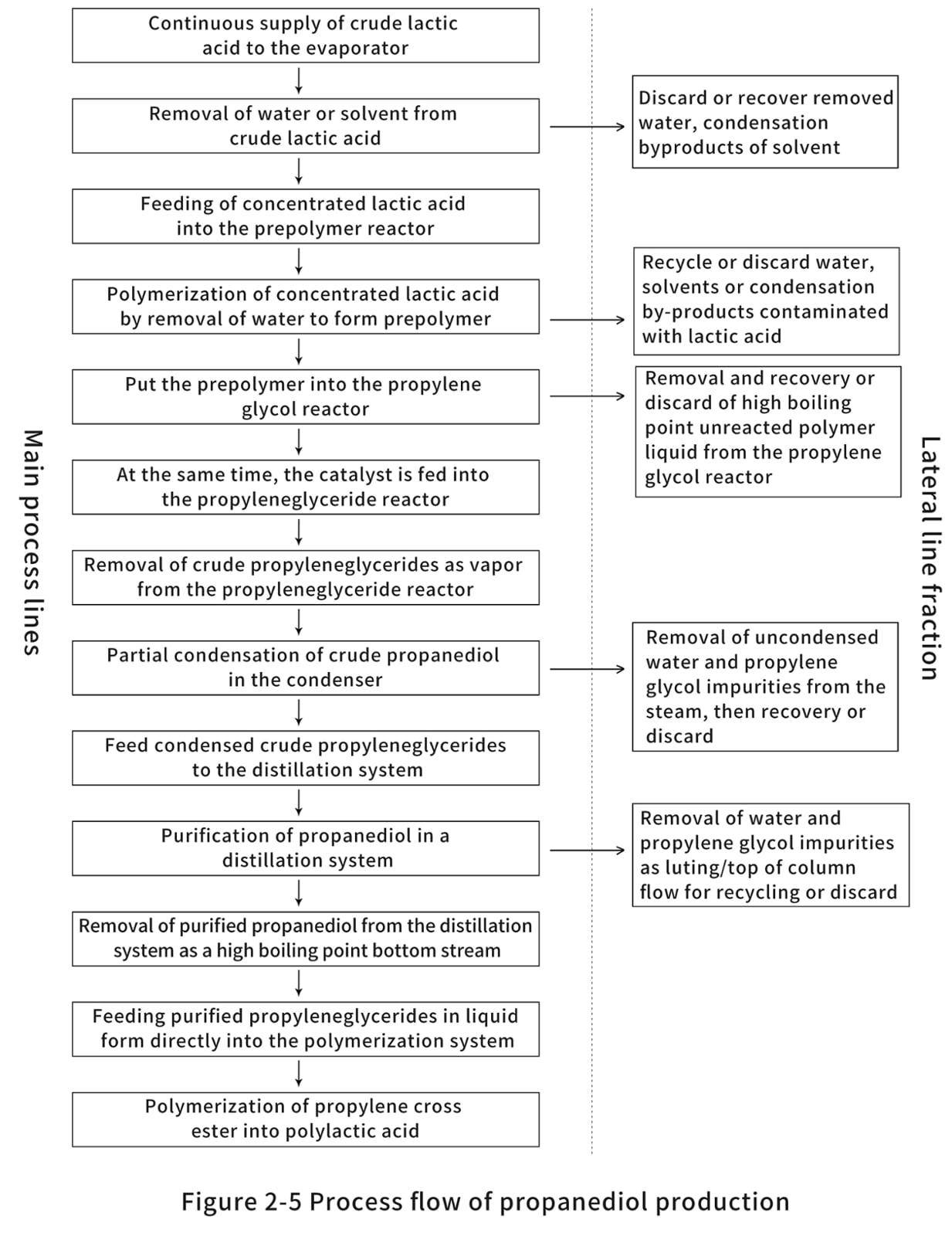
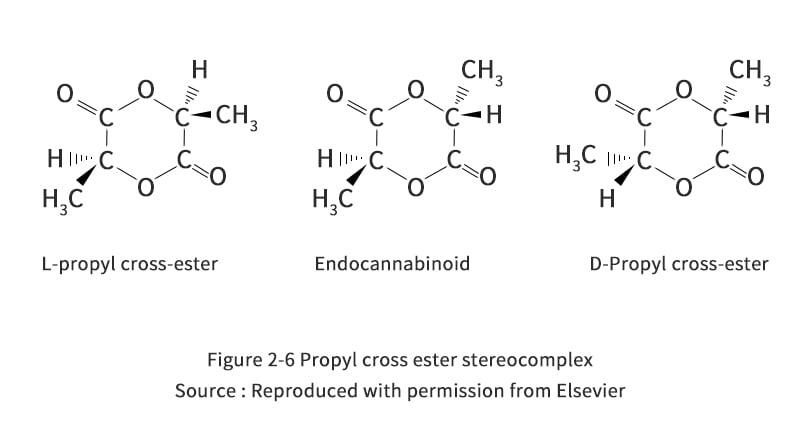
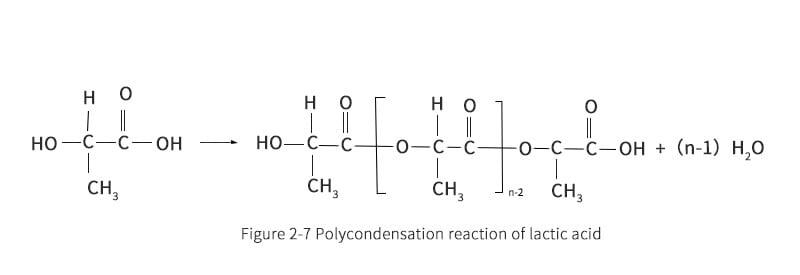
In the next stage, the concentrated LA is transferred to the prepolymer reactor. The prepolymer reactor is actually a second evaporator, which further removes water from the LA. At the same time, the polycondensation reaction is carried out to obtain PLA with molecular weights of 400 to 2500. When condensation polymerization of LA occurs, the alkoxy group reacts with the hydrogen from the hydroxyl cleavage of the nearest LA molecule. The remaining product is therefore a long LA bond and an excess of water molecules. To ensure that the right-hand side of the reaction path shown in reaction Figure 2-7 proceeds, it is important to remove water. During polymerization reactions, depolymerization reactions also occur due to the inherent equilibrium of the reaction system. Gruber et al. proposed an equilibrium reaction as shown in Figure 2-8.
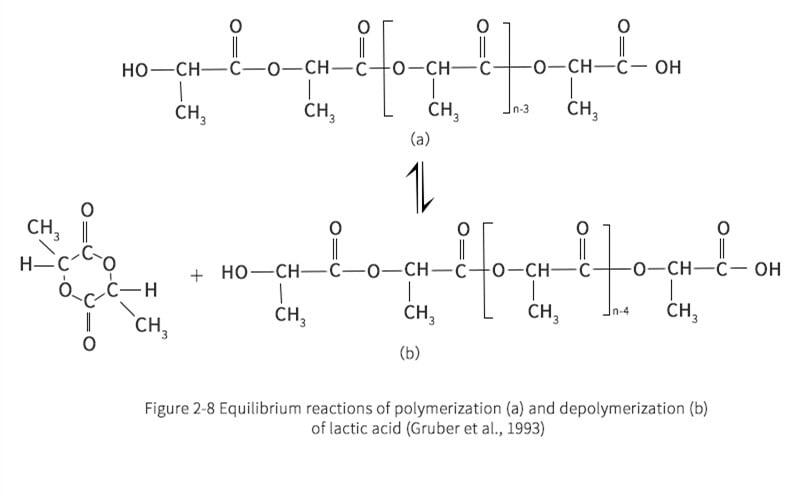
Gruber et al. claim that the prepolymer reactor can be designed as a single system, which can help facilitate LA feedstock concentration while polymerizing LA into oligomeric LA. However, the separation units in the evaporation and pre-polymerization stages are more controllable. To prevent raw material losses, LA recovery can be more efficient when water is separated from crude LA. Also, in the pre-polymerization stage, the reduced amount of high LA concentration helps to polymerize rather than depolymerize for better oligomeric LA yields. The oligomer LA, also previously known as prepolymer, is fed into the propylene-based reactor. Many suitable types of catalysts can enter the reactor at the same time as the prepolymer fluid. Commonly used catalysts are metal oxides, metal halides, metal dusts and organometallic compounds derived from carboxylic acids. According to the reaction scheme shown in Figure 2-8, the depolymerization reaction [Figure 2-8(b)] immediately reaches equilibrium. The reaction is carried out at high temperatures in order to gasify and continuously remove the crude propyl cross ester from the reactor, thus shifting the reactor to a depolymerization reaction. This follows the Le Chatelier principle, which states that the yield of the propyleneglyceride reactor is higher when the amount of propyleneglyceride is reduced in order to seek a reaction equilibrium in the propyleneglyceride reactor. However, unreacted long-chain PLA has a high boiling point and remains on the bottom when the reactor is cleaned. This product can be recycled into a prepolymer reactor or a propylene glycol reactor. The unreacted high molecular weight PLA undergoes an ester exchange reaction to form shorter oligomer chains, which is the source of the propyleneglycerides in the reactor. Utilizing this recirculation stream not only increases the recovery of valuable feedstock, but also helps to increase yields and reduce waste disposal costs.
As mentioned earlier, the composition of the produced propyleneglycol stereocomplex depends on the initial crude LA feed, the catalyst used and the process parameters (i.e. temperature and pressure). Thus, the crude propanediol vapor consists of a mixture of L-propanediol, D-propanediol, and racemic propanediol. Some low volatile products, such as water, LA and dimer LA, are also contained in this fluid. Partial condensers can be used to partially condense low boiling components such as water and LA prior to distillation. A conventional distillation column is used to separate the raw material into three components. The distillate or low boiling fraction at the top of the tower is water and LA, as well as other low molecular weight by-products from the prepolymer reactor and the propylene glycol reactor. The bottom fluid consists of a product that is less volatile than propylene glycol ester, such as an LA oligomer with more than three repeating units. In order to make the conversion of LA to propanediol more efficient, both the top and bottom products are recyclable. As a third component, propyleneglyceride is also extracted from the sideline fraction. The acceptable purity of propyleneglyceride is 75%; a high purity of propyleneglyceride is important for the formation of high quality PLA.
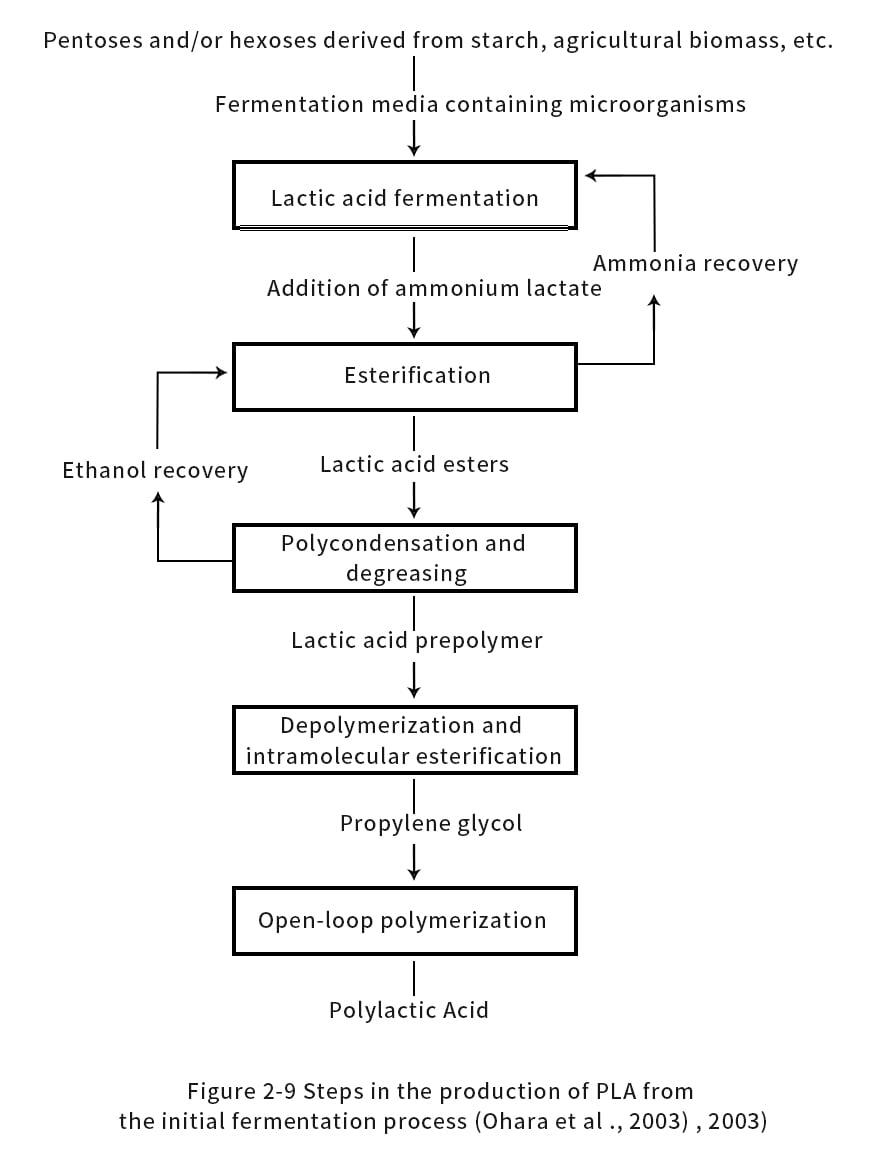
A more detailed method for synthesizing propyl cross-ester is disclosed in U.S. Patent 6569989 by Ohara et al. (Figure 2-9). LA was gradually condensed at 130-220°C in stages, and PLA prepolymer with molecular weights of 1000-3500 was obtained when the pressure was reduced to 5 mmHg at each stage. This multi-stage process can be further defined at different temperatures, where the first stage is at 135°C, the second at 150°C, the third at 160°C, the fourth at 180°C, and the fifth at 200°C. Metal-based catalysts were added to the reaction process to improve selectivity while shortening the reaction time. Since the polymerization and depolymerization take place simultaneously, similar catalysts are suitable for the production of propyleneglycerides. Therefore, metal catalysts can be added to prepare propyl cross-ester at reaction conditions of 200 °C and pressure of 5 mmHg. Depending on whether the LA or propanediol ester added to the reactor is fresh or crude, a mass ratio of 0.001% to 0.01% (mass fraction) is more appropriate to apply the catalyst.
2.3.2 Polymerization of propyleneglycolate
Most of the industry uses the ROP process of propylene glycol ester to prepare high molecular weight PLA. As shown in Figure 2-4, although DP appears to be the simplest reaction pathway for polymerizing monomer LA, PLA (molecular weight <5000) has relatively low yields and poor mechanical properties. Therefore, its application is limited. The ring polymerization is carried out in solvent systems initiated by anions and cations. It has the advantages of high reactivity and selectivity as well as low racemization and impurity levels. Methanetrifluoromethane sulfonic acid and methyltrifluoromethane sulfonic acid are cationic initiators that have been used to polymerize propyleneglycerides. This cationic ROP is carried out at low temperature (100°C) and the PLA products obtained are polymers with a single spin. The use of a primary alcohol salt such as potassium methanol as an anionic initiator yields racemic <5% PLA. however, anionic propyleneglyceride polymerization requires higher reaction temperatures, and reactivity is typically initiated at 120°C for weaker bases such as potassium benzoate and potassium phenol.
Although anionic and cationic initiation as described above has the advantage of producing low racemic PLA at lower temperatures, the reaction process needs to be carried out in a solvent system under dilute conditions to control its reactivity and sensitivity to the presence of impurities. The anionic and cationic initiators are also highly toxic. These aspects limit the application of anionic and cationic initiators in propyleneglyceride polymerization. In the large-scale PLA industry, metal-catalyzed methods are preferable for fast reaction rates and high yields in propyleneglyceride polymerization, and efficient catalysts are used only at low levels (<10 mg/kg), helping to ensure the safety of PLA in food packaging and biomedical applications. By using stannous octanoate (commonly known as tin octanoate), propyleneglyceride polymerization yields high molecular weights (<250,000), and the catalytic ROP reaction is also applicable to the polymerization of propyleneglyceride with other monomers such as ethyleneglyceride and ε-lactone.
Many catalyst systems are available for the polymerization of propanediol esters, including transition metals such as aluminum, zinc, tin and lanthanides. These metal oxides and complexes have varying degrees of conversion and high racemization. Tin or tinene (Sn) complexes are important for the native polymerization of propyleneglycolate, especially tin(II) bis-2-ethylhexanoate (also known as tin octanoate). Stannous octanoate is preferred due to its solubility in molten propanediol; therefore, it achieves a high conversion of >90% with high selectivity by producing a racemate of less than 1%. The high conversion rate facilitates good quality control in terms of mechanical and biodegradable properties. This is important for LA polymers for biomedical applications because the human body lacks LA, the enzyme that consumes D-lactate monomer after hydrolysis of the polymer to lactate, and only L-lactate can be consumed by living cells. At the same time, a large number of racemization interactions significantly affect the crystalline rearrangement structure and thus reduce the mechanical properties compared to single isomers.
The proposed scheme for the polymerization of propyleneglycolate with the addition of tin octanoate through the ligand-insertion mechanism is shown in Figure 2-10. The tin catalyst initiates the ring opening reaction by attacking the oxygen of the nearest double bond of the propyl cross ester. The hydroxyl and nucleophilic species react simultaneously with the ring-opening radicals, eventually forming water molecules as by-products to reach the steady state. The polymerization process yielded a low racemic mixture, high productivity and high molecular weight PLA. typical polymerization conditions were: 180-210°C, 100-1000 mg/kg of tin octanoate, 2-5 h conversion rate of 95%. The tin octanoate catalyst is also suitable for the copolymerization reaction of caprolactone and ethyl cross-ester, and the reaction scheme is shown in Figure 2-11. Residual catalysts from this polymerization process can cause unanticipated problems in dealing with degradation, hydrolysis or toxicity. Therefore, the addition of phosphoric or pyrophosphoric acid can render the catalyst inactive. The catalyst can also react with sulfuric acid and then be separated by precipitation. The level of catalyst in PLA or its copolymers should be reduced to 10mg/kg or less to ensure the quality of the application for the end user.
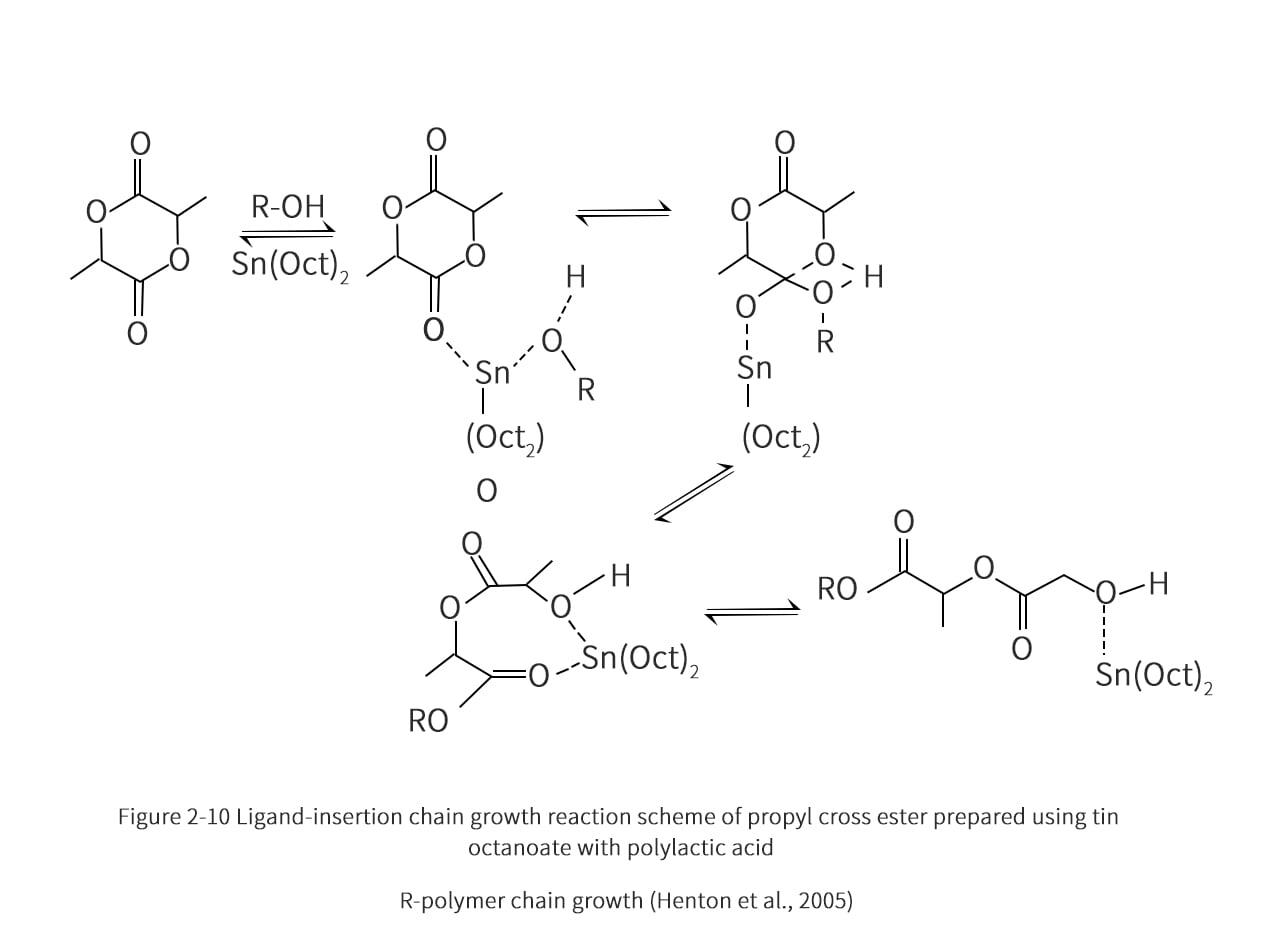
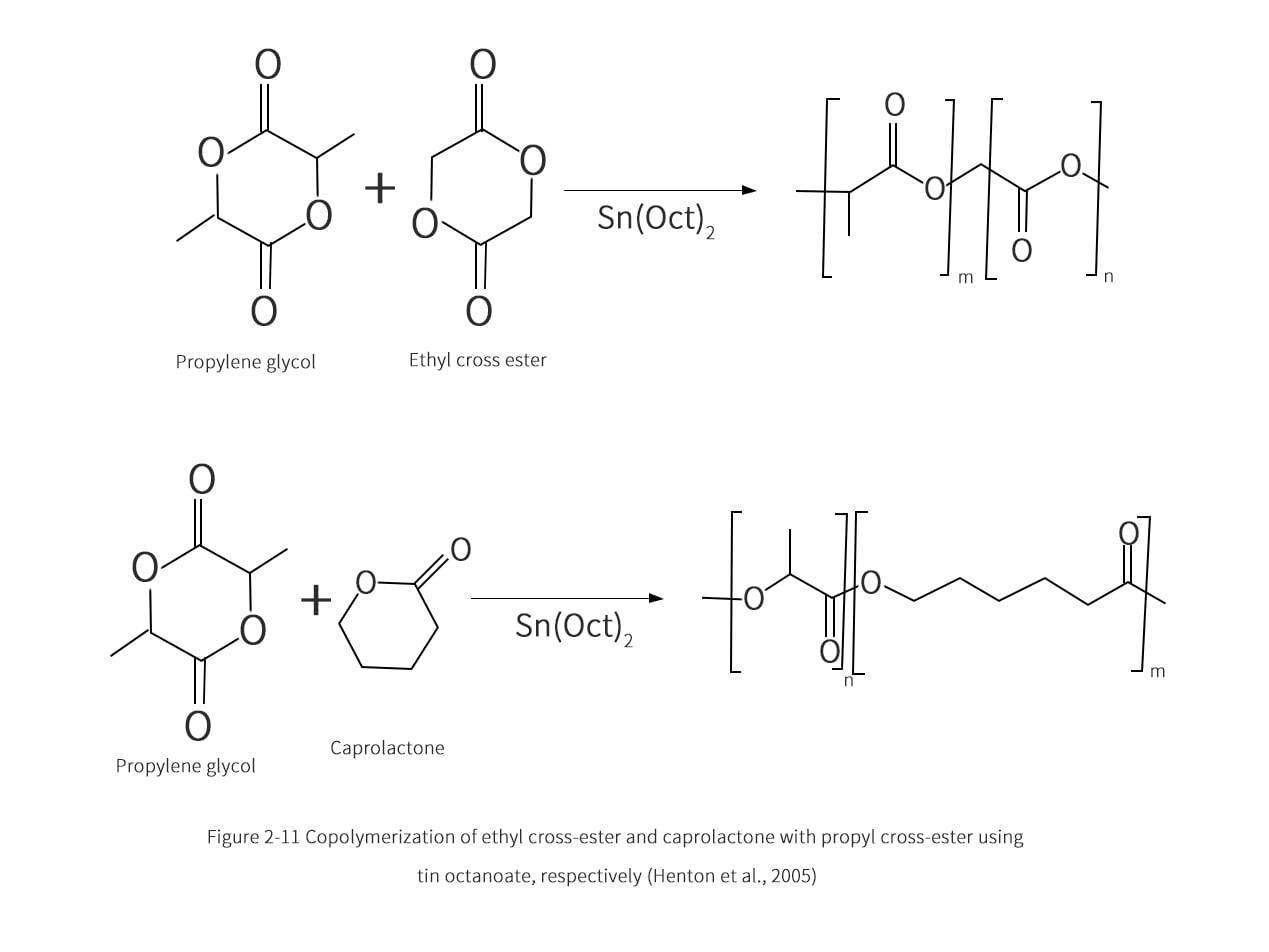
2.3.3 Propylene cross-ester copolymer
To improve the biocompatibility and good absorption time of implanted living tissues, propyl cross ester can be copolymerized with ethyl cross ester monomer. Typical applications of propylene-ethylene-glycolate copolymers, such as surgical sutures should contain greater than 80% ethylene-glycolate (by mass). This is because when less than 80% of the copolymer is ethylene cross ester, the degree of crystallinity is low and therefore lacks tensile strength and retention force in the application. The low content of ethylene cross in the copolymer is unfavorable because the predominance of propylene cross ester in the suture will reduce the absorption rate of the living tissue. The copolymerization process of propyl and ethyl cross esters is similar to the individual polymerization process of photoactive propyl cross esters. Stannous octanoate is also used as a catalyst for the copolymerization reaction, as shown in Figure 2-11. The two-step reaction process results in copolymers with high ethylene cross ester content. According to Okuzumi et al. the first stage is the polymerization of 65% to 75% of optically active propyl cross-ester with the remaining ethyl cross-ester monomer. In the second stage, a higher content of 80% to 90% of ethyl cross ester monomer was used in the co-polymerization reaction. Okuzumi et al. found that if reversal is attempted, the resulting propylene-ethyleneglycolate copolymer has a low molecular weight and forms an amorphous polymer, making it unsuitable for surgical sutures, which require high-strength fibers. Table 2-4 summarizes the results of this observation.
Table 2-4 Tensile strength of propylene-ethyleneglyceride copolymer
Mass fraction of propyl cross-ester-ethyl cross-ester
First stage copolymerization | Second stage copolymerization | Final copolymer composition | Tensile strength/103psi |
40/60 | 0 | 40/60 | 53 |
70/30 | 12/88 | 35/65 | 64 |
70/30 | 12/88 | 40/60 | 67 |
70/30 | 12/88 | 45/55 | 71 |
87/13 | 12/88 | 35/65 | 60 |
87/13 | 12/88 | 50/50 | 58 |
78/22 | 12/88 | 35/65 | 71 |
78/22 | 12/88 | 45/55 | 63 |
78/22 | 18/72 | 50/50 | 58 |
Note:1MPa=145psi。
Propyl cross-ester is also copolymerized with ε-lactone monomers to produce biomaterials for the manufacture of surgical implants and drug carriers. The copolymerization reaction route of propylene-caprolactone is similar to that of propylene-ethylene-caprolactone, and a random copolymer containing 55% to 70% (molar fraction) of propylene-caprolactone and 30% to 45% (molar fraction) of already lactone is preferred for use as a drug carrier.
Although the above-mentioned copolymers of propylene-ethylene-caprolactone are suitable for the manufacture of medical devices with excellent properties such as high strength, high stiffness, and maintenance of fracture strength over time, copolymerization of propylene-caprolactone with dioxane monomers can improve the tensile properties of propylene-caprolactone in toughening applications such as absorbable medical devices, tissue scaffold foams, and hemostatic barriers.
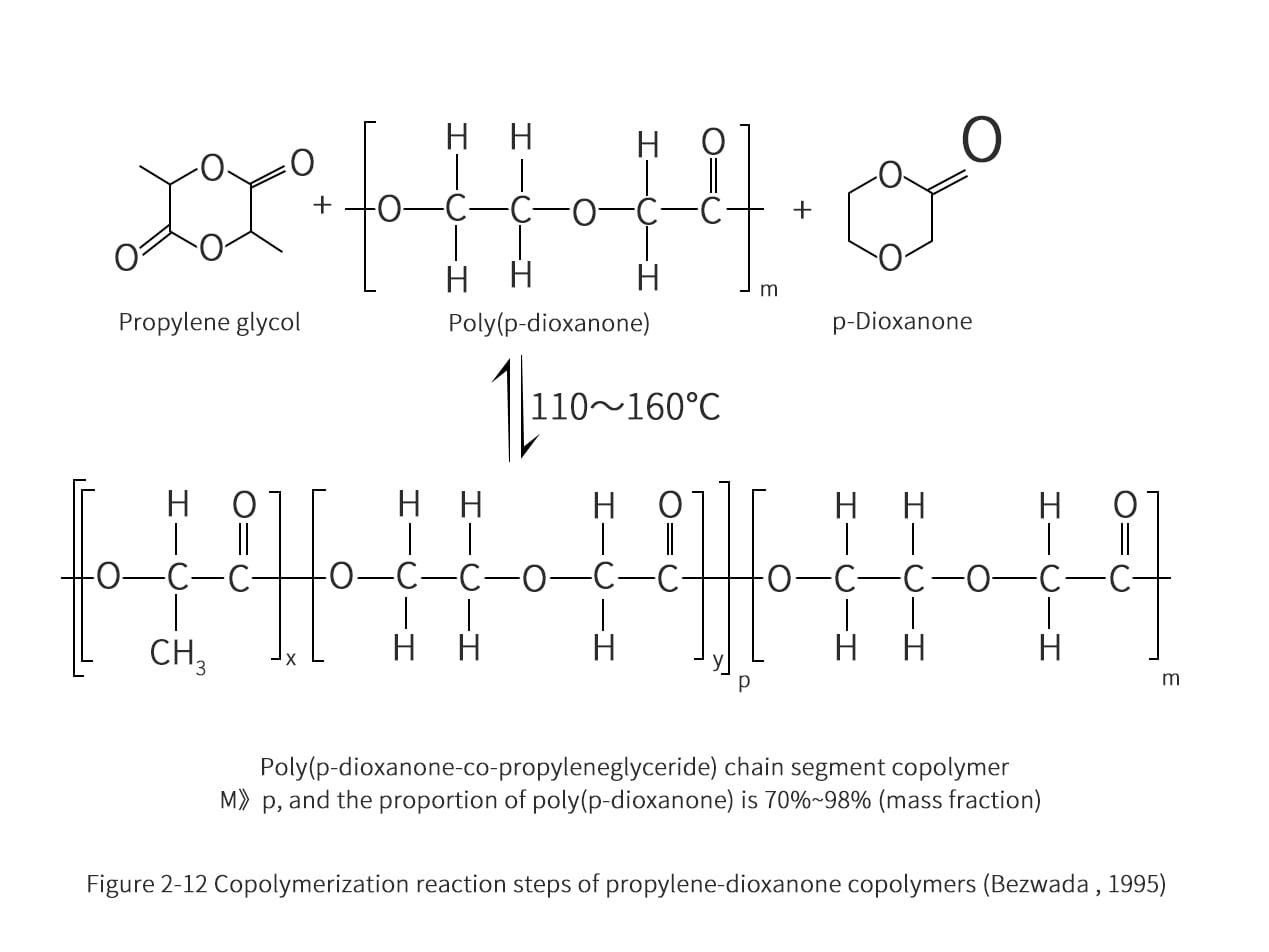
The production of propylene-dioxane copolymer was also performed in two steps (Figure 2-12). Initially, the propanediol ester was reacted with a small amount of p-dioxane monomer at 100-130°C for 4-8 h. Subsequently, the temperature was increased to 160-190°C for 1-4 h to allow further copolymerization of the propanediol ester with the long-chain poly(p-dioxane) prepared in the first step. The final product is a high strength, high tenacity elastomeric biopolymer containing 30% to 50% (molar fraction) of propyleneglycolate. The use of low toxicity and highly selective tin octanoate in the copolymerization process yields high molecular weight mosanone copolymers with molecular weights of 60,000 to 150,000.
2.3.4 Quality control
The mass production of PLA is most commonly used in the manufacture of everyday consumer products such as packaging or bottles that come into contact with food. For these PLA products, quality control no longer limits mechanical properties as it does for general purpose polymers (polyethylene, polypropylene, polystyrene, etc.). However, manufacturers need to carefully classify the amount of propanediol in the final product, especially the amount of D-lactic acid. As the largest manufacturer of PLA, NatureWorks has established a standard test procedure for companies that manufacture with its Ingeo products as shown in Figure 2-12. Although these tests were developed by NatureWorks, their application is not limited and they can be used widely throughout the PLA industry.
2.3.5 Determination of residual propanediol in polylactic acid (NatureWorks L.L.C., 2010b)
The composition of propyleneglycerides in PLA was determined by gas chromatography (GC) with a flame ionization detector (FID). This GC/FID method can only detect residual propoxur in the range of 0.1% to 5% (mass fraction). Although the detection range was narrow, the propanediol monomer found in PLA remained within the concentration range of 3% (mass fraction) at 180 °C, and the concentration could be further reduced to 0.3% (mass fraction) after PLA liquefaction. As mentioned earlier, the propanediol monomer consists of three stereoisomers: L-propanediol, D-propanediol, and racemic propanediol. The GC method can only detect two propyleneglyceride peaks (detected in a single peak) of endocyclic propyleneglyceride and D-propyleneglyceride or L-propyleneglyceride. The endocyclic propanediol has the earliest elution peak, while the subsequent elution peaks indicate the coexistence of D-propanediol and L-propanediol. The GC/FID method starts with the preparation of four solutions, i.e. 1 internal standard master batch, 2 propyleneglyceride standard master batch, 3 propyleneglyceride working standard solution, and 4 PLA sample solution. Dichloromethane is the solvent used to dissolve the PLA and release the free propyleneglyceride. The free propanediol is retained in dichloromethane while an excess of cyclohexane is added to precipitate PLA. The supernatant is then filtered and injected into the GC, and finally detected by FID. The choice of GC injection temperature is critical and must be 200°C to avoid the possibility of propyleneglyceride conversion due to the presence of low molecular weight LA oligomers in the supernatant.


2.3.6 Determination of D-lactic acid content in polylactic acid
(8) It is important to evaluate the amount of D-lactic acid, especially if the PLA product will be in contact with food or as a biological implant. The allowable daily intake of D-lactate for adults is 100 mg/kg, and D-lactate should not be contained in foods for infants. Chiral gas chromatography with a hydrogen flame ionization detector was used to detect the residues of D-propyl cross-ester and L-lactic acid in PLA samples. In this method, the sample is first hydrolyzed in methanolic potassium hydroxide and then acidified under strong acid to catalyze the esterification reaction. Then, dichloromethane and water were added to the acidification solution to separate the organic layer of methyl lactate enantiomer dissolved in dichloromethane at the bottom and the upper non-organic water layer into a bilayer. The underlying organic layer was collected and analyzed using a GC-FID system. The procedure for preparing PLA samples for testing was as follows.
1. Dissolve the PLA samples in potassium hydroxide methanol solution at 65°C;
2. Sulfuric acid was added to the sample solution and heated again to 65°C;
3. Add deionized water and methylene chloride;
4. Splitting the liquid sample into two layers;
5. The substrates of the samples were mapped and analyzed by GC-FID.
It is recommended to use an Agilent J&W CycloSil-B gas chromatographic column in DB-1701- stationary phase with 30% heptane (2,3-di-O-methyl-6-O-tert-butyldimethylsilyl)-β-cyclodextrin to separate the methyl lactate enantiomers. β-cyclodextrin is suitable for chiral splitting because its cyclic oligosaccharide units form inclusion complexes with different equilibrium constants with methyl lactate enantiomers, which can be easily separated by gas chromatography. The method has a wide detection range of 0.05% to 50% of D-lactate in PLA.
(1) The relative percentages of D-lactate and L-lactate enantiomers present in PLA were calculated as follows:

(2) The prescribed GC-FIB test method has an accuracy of less than 1% with respect to standard deviation for the determination of D-lactate in PLA.
2.4 Catalysts for PLA polymerization reactions
2.4.1 Direct polycondensation route
It is well known that there are two common synthetic routes for the preparation of PLA, namely, the ROP method for propyleneglycerides and the DP method for LA. Although the polymerization reaction for the production of PLA from LA is a one-step polymerization reaction, its application in industrial production is limited by the poor control of molecular weight in the DP process. Overall, although the ROP route requires additional reaction steps, the polymerization process is easy to control due to its ease of use. It has been commonly used in the industrial production of PLA. In the past decades, there have been many studies to improve DP by obtaining high molecular weight PLA polymers. a new synthetic method was developed by Moon et al. to increase the molecular weight of PLA by introducing the melt polymerization reaction of LA prior to solid phase polycondensation. They successfully prepared poly(lactic acid) polymers with molecular weight over 100,000 by melt polymerization and solid-phase polycondensation using stannous chloride and p-toluenesulfonic acid (SnCl2/p-TSA) system as catalysts. PLA polymers are one of the most important polymers in the biomedical and food packaging industries due to their biological origin, their biodegradability, and the fact that they are environmentally absorbable through conversion to carbon dioxide and water. However, its poor mechanical properties such as toughness and thermal stability limit its application in various industries. Wu et al. prepared poly-L-lactic acid/silicon dioxide (SiO2) nanocomposites by in situ melt polymerization of L-lactic acid in acidic silica sols. Stannous chloride (SnCl2-2H2O) and p-toluenesulfonic acid (TSA) were used as binary catalysts to induce L-lactic acid activity during in situ melt polymerization. Silica nanoparticles were chemically grafted onto L-lactide oligomers using an in situ melt polymerization method. Furthermore, it is known that the use of SnCl2-p-TSA systems as catalysts for the production of poly(L-lactide) is dangerous because SnCl2-p-TSA tends to remain in the produced poly(L-lactide). SnCl2-p-TSA catalyst is toxic and difficult to remove from the produced poly(L-lactic acid), leading to its limited application in the biomedical and food packaging industries. Ren et al. replaced SnCl2,p-TSA with Amberlyst-15, a green catalyst macroporous resin, as a catalyst in the melt polycondensation reaction of L-lactic acid. Unlike the SnCl2-p-TSA catalyst system, Amberlyst-15 is a non-toxic solid acid that can be easily separated from the poly(L-lactic acid) prepolymer by a simple filtration method. The non-toxic Amberlyst-15 catalyst has good catalytic properties, is readily available, inexpensive, and can be reused three times during the reaction with nearly identical catalytic activity. The molecular weight of the poly(L-lactide) prepolymer produced with Amberlyst-15 reached 46,000. this finding also suggests that the Ambelyst-15 catalyst is not only safer than the SnCl2-p-TSA catalyst system, but also exhibits very similar catalytic activity in the melt polymerization of L-lactide to poly(L-lactide) prepolymer.
Ajioka et al. found that organosulfonic acid was often used as an effective catalyst in the condensation of L-lactic acid solution for the preparation of poly-L-lactic acid with comparable molecular weight. Similar work was carried out by Takenaka et al. to prepare high molecular weight PLA by melt polymerization and solid phase polymerization reaction using sodium dodecylbenzene sulfonate as a catalyst. Using 0.7% (mass fraction) sodium dodecylbenzene sulfonate as the catalyst, LA was first subjected to ontogenic melt polymerization at an annealing temperature of 80-110°C to obtain PLA prepolymer with molecular weight greater than 3000. The prepared poly(L-lactic acid) prepolymer was subjected to a solid-state polycondensation process at 140 °C to prepare poly(L-lactic acid) polymers with molecular weights up to 115,000. They also found that, among all aromatic sulfonic acids, sodium dodecylbenzenesulfonate was the best catalyst to provide and maintain catalytic activity throughout the solid-phase polycondensation process with long reaction times. In addition, the decomposition temperature of dodecylbenzenesulfonic acid is the highest (over 200°C) while the decomposition temperature of other sulfonic acids is around 150°C. The long condensation reaction time leads to the decomposition of other sulfonic acids and loss of their initial catalytic activity. Bai and Lei investigated the effect of different types of organic anhydride catalysts, such as acid anhydride, maleic anhydride, homophthalic dianhydride, and phthalic anhydride, on the polycondensation of LA for the preparation of PLA. The results showed that the application of these organic anhydride catalysts in the native polycondensation reaction played a positive role in improving the yield of PLA products by obtaining poly(D,L-lactide) polymers with average molecular weights in the range of 70,000 to 90,000. Huang et al. successfully synthesized poly(L-lactic acid) and poly(D-lactic acid) with molecular weights up to 120,000 and 100,000, respectively, using biological creatinine as a catalyst by melt polycondensation and solid-phase polycondensation, as shown in Figure 2-13. Poly(L-lactic acid) and poly(D-lactic acid) of 97.8% to 99.4% isocratic degree were prepared by melt polycondensation and solid-phase polycondensation, and the riser chemistry of PLA was controlled during the whole polymerization process. In addition, the degradation temperature of PLA synthesized by molten solid-phase polycondensation catalyzed by biogenic creatinine was also at least 100 °C higher than that of PLA synthesized by SnCl2-H2O.

Pivsa Art et al. synthesized poly(D-lactide) by two-step direct polymerization of D-lactide using 2-NSA as catalyst. First, poly(D-lactide) prepolymer was prepared by melting polymerization of D-lactide through esterification reaction using 2-NSA as catalyst. The prepared PLA prepolymer was further subjected to solid phase polycondensation at high temperature and decreasing pressure. They successfully synthesized PLA to meet the requirements of thermal properties (decomposition temperature of 255°C) and medium average molecular weight of 33,300. On the other hand, Achmad et al. synthesized poly(L-lactic acid) by DP reaction under vacuum conditions without catalyst, solvent and initiator. They claim that the non-catalytic direct polymerization of L-lactic acid is a secondary reaction mechanism. This is because L-lactic acid itself is a strong acid, and polymerization under vacuum conditions at a polymerization temperature of 150-250°C.
In addition, the activation energy of direct polymerization without catalyst was found to be higher than that of ring-opening polymerization with catalyst.
2.4.2 Open-loop aggregation route
In the production of PLA, the propylene-crosslinked ester ring-opening polymerization reaction route is a reactive polymerization method of current industrial interest due to its better reaction control in the production of high molecular weight PLA. The industrial production of high molecular weight PLA is usually based on the ROP of L,L-propyl cross-ester reaction with stannous sulphoxide [Sn(Oct)2] as catalyst. Stannous octanoate is the most commonly used catalyst for the ROP of L, L-propyl cross-ester in PLA industrial production. This is due to the high polymerization rate of propyleneglycolate, its good activity in ROP, and its approval by the Food and Drug Administration (FDA) for use in the biomedical industry. Zhang et al. (1994) investigated the role of hydroxyl and carboxylic acids such as ethanol as co-catalysts in the ROP reaction of propyl cross ester using stannous octanoate as catalyst. They found that the preparation of PLA from stannous octanoate-catalyzed ROP with different co-initiators may affect the molecular chains of PLA, producing linear or branched polymer chains with different molecular weights. They also found that the reaction between stannous octanoate and alcohols can produce alkoxylated stannous, which can initiate the ROP reaction by insertion into the polymer chain. The initiation mechanism of the ring-opening reaction is known as the alcohol salt initiation mechanism. The reaction of alcohols with stannous octanoate can significantly affect the ROP reaction through different reaction stages such as initiator formation, chain transfer and ester exchange reaction. On the other hand, the application of carboxylic acid substances with stannous octanoate as catalyst affects the ROP reaction through deactivation reactions, and Zhang et al. also found that the use of ethanol and carboxylic acid substances in ROP containing stannous octanoate significantly reduced the final molecular weight of the resulting PLA. However, the use of alcohols in the ROP reaction catalyzed by stannous octanoate improves the yield of PLA.
In the study of Kowalski et al. they prepared polylactic acid from L,L-propyl cross-ester by alcohol salt initiation mechanism using stannous octanoate as catalyst and butanol (BuOH) as initiator. They performed the ROP of L. L-propyl cross-ester in THF below 100 °C and made the initial reaction of stannous octanoate with BuOH by forming polymer chains with Oct-SnO end groups such as OctSnOBu and OctSnOBu compounds. These compounds are actually initiators of L,L-propyl cross-ester ROP. Zhang et al. and Kowalski et al. also investigated the rate of L, L-propyleneglyceride polymerization induced by ethanolic substances as co-initiators to form PLA in propyleneglyceride ROP with stannous octanoate as catalyst compared to the presence of trace amounts of water, as shown in Table 2-5.
Table 2-5 Effect of different types of initiators on the type and molecular weight of PLA formed and polymerization reactivity
Co-initiators | Type of polypropylene cross ester formed | Polymerization reactivity | Molecular weight and polymerization time |
Trace water | Linear PLA | Minimum activity | Low molecular weight |
Mono- and difunctional alcohols ·1-Dodecanol ·1,4-Butanediol ·2-Hydroxy methyl propionate ethyl ester |
Linear PLA |
Higher than polyol and trace trace water
|
Increase molecular weight and polymerization rate |
Polyol ·Butanol ·Pentaerythritol ·2,2-Hydroxymethyl-1,3-propanediol Diol ·Bis(trimethylolpropane) or DTMP ·Glycerin ·Linear polyglycidylic acid (with 25 hydroxyl groups) |
Branched chain polylactic acid Four-armed polylactic acid Four or six long branch chains Dobby Star Branched Poly Lactic Acid |
①Higher than trace water, but lower than mono- and di-functional groups ②The relative reactivity of hydroxyl groups decreases with the increase in the number of hydroxyl groups | Increase in molecular weight; however, longer polymerization times may reduce molecular weight compared to linear PLA due to faster degradation kinetics and higher rates of unstable bond cleavage along the polymer backbone than linear PLA |
According to the summary of Yu et al. and Averianov et al. the choice of different types of alcohol substances, such as mono-hydroxy, bi-functional hydroxy alcohols or polyols (>2 via groups), had a significant effect on the molecular structure of PLA generated by ROP with stannous octanoate as catalyst as shown in Table 2-5.
Referring to Table 2-5, linear PLA can be synthesized using mono- and bifunctional alcohol substances such as 1-dodecanol and 1,4-butanediol as co-initiators or co-catalysts.
It was also found that the application of mono- and di-hydroxy alcohol substances leads to higher polymerization reactivity than water and polyol substances. On the other hand, branched PLA can be synthesized in the ROP process of propyleneglycerides by using polyols such as butanol and pentaerythritol as co-catalysts. According to Korho-nen et al. and Karidi et al. polyols with 25 hydroxyl groups, such as pentaerythritol, bis(trimethylolpropane) (DTMP), glycerol, and polyglycidol, yielded dobby and 4 or 6 long-chain branched polylactic acid. They also showed that the relative reactivity of the hydroxyl groups of the alcoholic substances used decreased significantly as the number of hydroxyl groups increased. In addition, they found that the longer the polymerization time of the propylene-cross-ester ROP catalyzed by stannous octanoate with polyols as co-catalysts could significantly reduce the molecular weight of the resulting PLA due to the faster degradation of the branched PLA than the linear PLA. This can be attributed to the high rate of unstable bond breakage along the main chain of the branched PLA polymer, which degrades the obtained PLA polymer.
In order to produce PLA with higher conversion and molecular weight, a lot of research has been done on the synthesis of new catalysts for propylene-based ROPs. Routaray et al. successfully synthesized copper(II) complexes using the ONNO tetradentate ligand N, N'-bis (2-hydroxy-3-methoxybenzaldehyde) -benzene-1,2-diamine (Cu-HMBBD) as a carrier, which was used as a catalyst to induce the reaction of propanediol ROP to produce PLA. They further investigated the effects of different types of co-initiators, such as benzyl alcohol (BnOH), CH2Cl2, toluene and THF on ROP with Cu-HMBBD as catalyst. With BnOH as co-initiator and Cu-HMBBD as catalyst, the conversion of the final product reached 94.6% at a peak molecular weight of 28600. In addition, the use of BnOH as a co-initiator also resulted in a reduction of the polymerization time from 24 h to 11 h due to the good interaction between the polar solvent of BnOH and Cu-HMBBD in the polymerization reaction. Kang et al. also synthesized two iron complexes, i.e., dimeric iron(III) complexes containing acetylacetonate (acac) ligand and tridentate as a catalyst for propyl cross ester ROP chiral Schiff base ligand to form polylactide. Compared with the dimeric iron(III) complex containing acetylacetonate (acac) ligand, the conversion increased from 22% to 97% and the molecular weight of the final product increased from 2400 to 7300 when the tridentate chiral Schiff base ligand was used as a catalyst in the ROP process of propyl cross-ester. Li et al. synthesized four different aluminum complexes, namely [2-(anilino)cycloheptatrienone] AIMe2, {2-[2-(phenoxy)anilino]cycloheptatrienone} AIMe2, {2-[2-(2,6-diisopropylphenoxy)anilino]cycloheptatrienone} AIMe2 and {2-[2-(phenylthio)anilino]cycloheptatrienone} AlMe2. The ROP reaction of propyl cross-ester was carried out in toluene at 80 ℃ using the synthesized aluminum complex as catalyst and benzyl alcohol as co-initiator. Compared to other aluminum complexes, the lowest conversion (52%) was found with [2-(phenylamino)terephthalic acid] AIM2, as catalyst in the polymerization reaction, while the highest conversion and molecular weight (92% and 14100, respectively) were obtained with 2-[2(phenoxy)aniline]terephthalic acid AIM2. Schmitz et al. also synthesized seven aluminum ion-pair complexes by reacting NNO Schiff base ketimine with different substituents of tris(2,6-dimethylphenoxy) as catalysts for L-propyl cross-ester and racemic propyl cross-ester ROPs to prepare poly(lactic acid).Luo et al. used Zn(NO3)2.6H2O, 1,4-diazabicyclo[2,2,2 ]octane (H2BDC) and 1,4-benzenedicarboxylic acid (DABCO) to synthesize four different types of metal-organic backbone MDABCO (e.g.: ZnDABCO). Luo et al. have demonstrated that MDABCO catalysts are highly active in the ROP for the production of L-propyl cross-ester of PLA without co-emergent, especially the highest ZnDAB- CO conversion and molecular weight. Using LA as co-initiator, they further investigated the effect of ZnDAB- CO as catalyst on L-propyl cross-ester ROP. It was found that LA used as co-initiator and ZnDABCO used as catalyst can greatly improve the polymerization activity of the system. CaoVilla et al. (2018) successfully synthesized four different types of zinc bipyrrolyl imine complexes (Zn1, Zn2, Zn3, Zn4) by the reaction of ZnEt2 with different types of pyrrolyl imine ligands. They investigated the conversion of these synthesized zinc-bipyrrolidinimine complexes as catalysts in the propyleneglyceride ROP in terms of propyleneglyceride conversion and end product molecular weight. The results showed that the conversion of propyleneglycerides could reach 99% when BnOH was added and Zn1 was used as the catalyst at a polymerization temperature and time of 80°C and 96h. However, when the polymerization temperature was increased from 80°C to 130°C for 1h, the conversion of endoester decreased significantly. Among all the synthesized zinc bipyrrolyl imide complexes, the ROP reaction of propyl cross ester utilized Zn2 and Zn3 as catalysts to obtain maximum molecular weights up to 19,330-19,390 and conversions of 94%-97% at a polymerization temperature and time of 130 °C and 1 h with BnOH as co-initiator. Caovilla et al. also found that Zn1, Zn2, Zn3 and Zn4 catalyzed the ROP reaction of propyleneglycolate to produce PLA with faster and better controlled polymerization when excess benzyl alcohol was used as co-initiator.
2.5 Conclusion
PLA is produced from the starting material LA, which is obtained by fermentation of carbohydrates. PLA can be produced by DP method or propylene glycol ring-opening polymerization. Of these two methods, propylene-crosslinked ester ring-opening polymerization remains the most widely used because of the higher yield and lower toxicity of this method. In addition, ring-opening polymerization of propyleneglycerides is suitable for copolymerization of propyleneglycerides with lactones, ethyleneglycerides or dioxane ketones, where the propyleneglycerides and D-lactic acid in PLA need to be measured to avoid excess. The choice of catalyst is also important for both polymerization methods (ROP of DP or propylene-crossing). Suitable catalysts can effectively induce the conversion of LA or propyleneglycerides to PLA and increase the molecular weight of the resulting PLA. In general, understanding the production and quality control of PLA is very helpful to ensure the long-term development of PLA.
3. Thermal properties of polylactic acid
3.1 Introduction
Poly(lactic acid) (PLA) is a biodegradable aliphatic semi-crystalline polyester that starts with a direct condensation reaction of its monomer lactic acid as an oligomer followed by ring-opening polymerization of a cyclopropyl cross-ester dimer. The lactic acid optical monomer consists of L-lactic acid and D-lactic acid, as shown in Figure 3-1. From these two optical monomers, lactic acid oligomers can form three possible stereomorphs of propanediol: L-propanediol, D-propanediol and racemic propanediol (also known as D,L-propanediol; see Figure 3-2. The purified L-propyl cross-ester, D-propyl cross-ester and racemic propyl cross-ester dimers were converted to the corresponding high molecular weight polyesters by catalytic ring-opening polymerization. The stereochemical composition of PLA has a significant effect on its melting point, crystallization rate, degree of crystallization, and mechanical properties. This chapter discusses the thermal properties of PLA and PLA-based composites, including heat capacity, thermal transition, thermal decomposition, and crystallization.
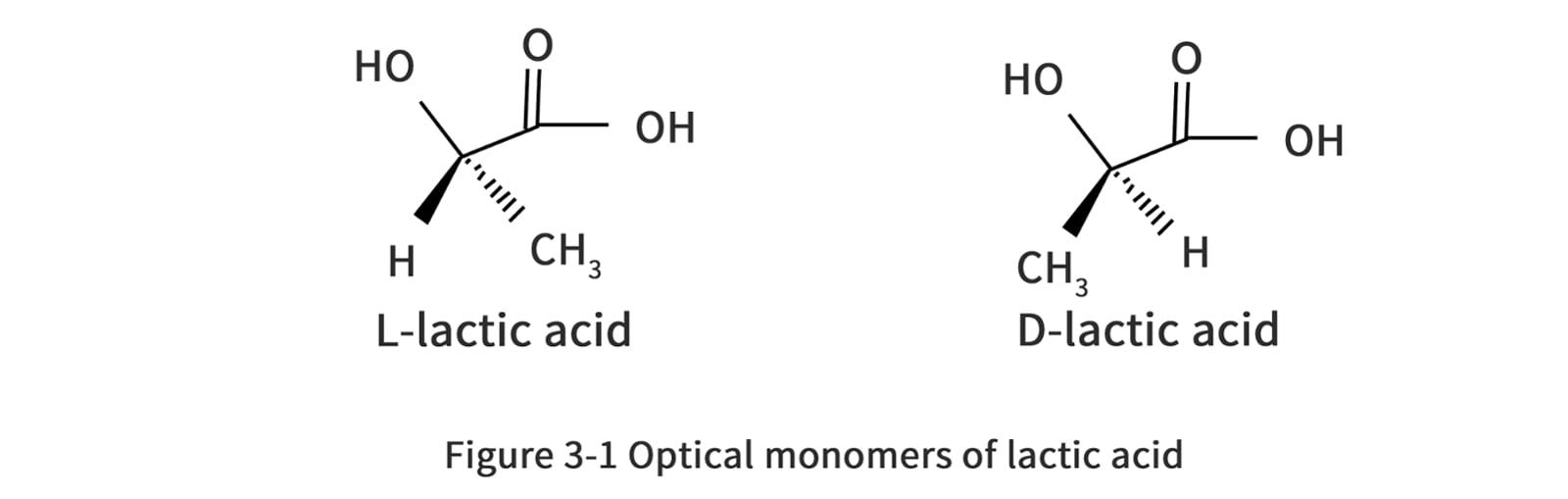
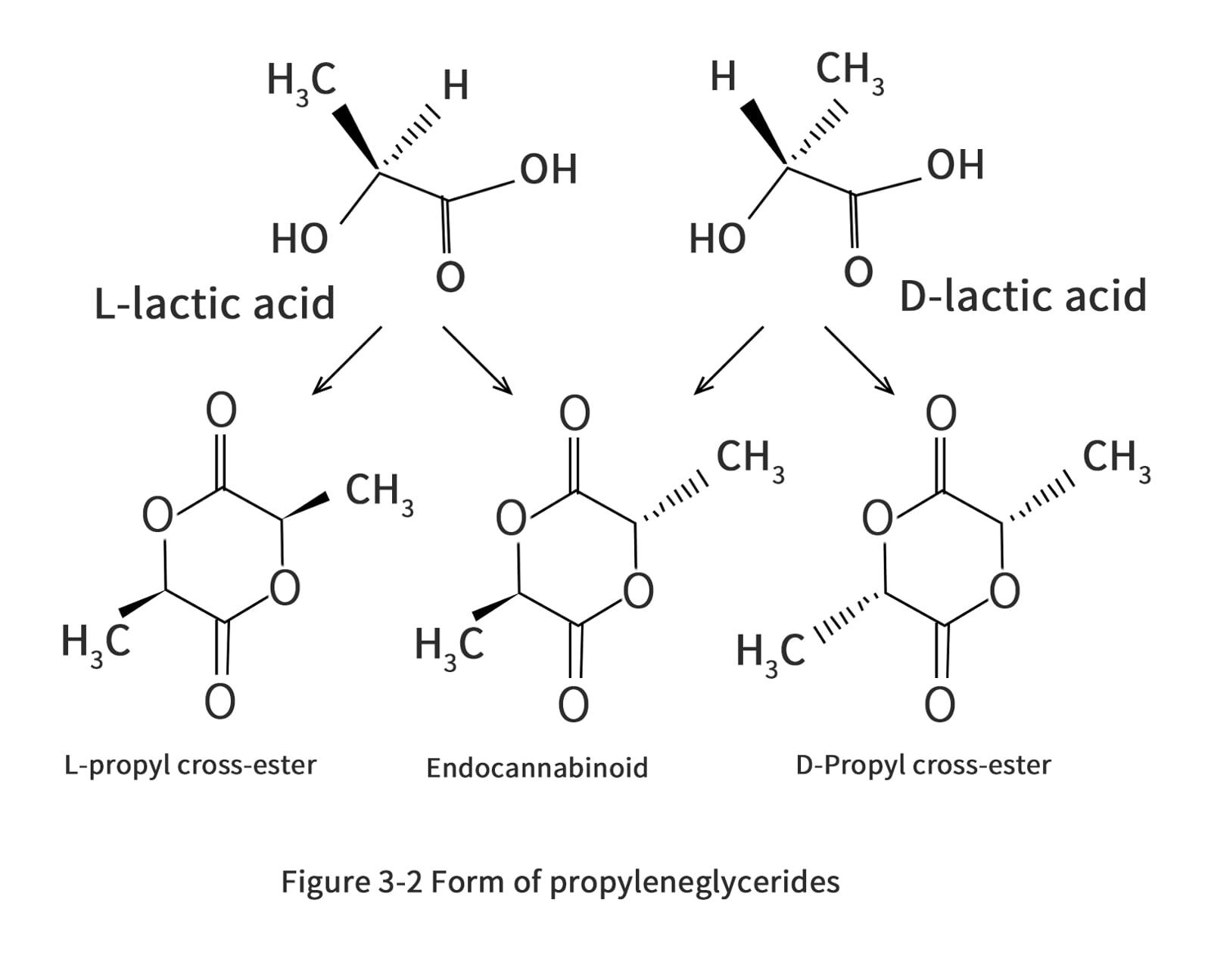
The thermal properties of PLA are usually determined by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and dynamic mechanical analysis. The crystallization behavior, crystallinity and thermal properties of PLA depend on the molecular weight of the polymer, polymerization conditions, thermal history, purity, etc. Achmad et al. reported that poly(L-propyleneglycolate) (PLLA) and poly(D-propyleneglycolate) (PDLA) are semi-crystalline polymers with a melting point of about 180°C, while the copolymer poly(D,L-propyleneglycolate) (PDLLA) is an amorphous material with a glass transition temperature of only 50 to 57°C. As seen in Table 3-1, different propyleneglycolate isomers have significant effects on the molecular weight (Mn, glass transition temperature (Tg), melting temperature (Tm), enthalpy and crystallization temperature (Tc) of PLA. It can be observed that the Tg and butm of PLA increase with the increase of Mn regardless of the monomeric isomer type L or D. When the temperature is below Tg, information about Tg is important for polymers because the material is essentially frozen, so no large-scale molecular motion is possible, whereas if the temperature is above Tg, molecular motion can occur on the scale of its repeating units (e.g., individual units in a polymer), making it "soft" or "elastic". In other words, the Tg of a polymer is related to its processability and service temperature. PLA with low Tg is not suitable for hot water because the material will soften and deform. PLA with low Tg is not suitable for hot water because the material will soften and deform.
Table 3-1 Effect of isomers on the thermal properties of PLA(Ahmed et al., 2009)
Type of isomers | Mn/×103 | Mw/×Mn | Tg/×℃ | Tm/×℃ | ΔHm/(J/g) | Tc/℃ | ΔHc/(J/g) |
L | 4.7 | 1.09 | 45.6 | 157.8 | 55.5 | 98.3 | 47.8 |
D,L | 4.3 | 1.90 | 44.7 |
|
|
|
|
L | 7.0 | 1.09 | 67.9 | 159.9 | 58.8 | 108.3 | 48.3 |
D,L | 7.3 | 1.16 | 44.1 |
|
|
|
|
D | 13.8 | 1.19 | 65.7 | 170.3 | 67.0 | 107.6 | 52.4 |
L | 14.0 | 1.12 | 66.8 | 173.3 | 61.0 | 110.3 | 48.1 |
D | 16.5 | 1.20 | 69.1 | 173.5 | 64.6 | 109.0 | 51.6 |
L | 16.8 | 1.32 | 58.6 | 173.4 | 61.4 | 105.0 | 38.1 |
However, PDLLA copolymers made from the polymerization of L-lactic acid and D-lactic acid were unable to detect melting and crystallization even at high molecular weights, due to the atactic configuration affecting the microstructural rearrangement upon cooling. Meanwhile, after comparing the L isomers with Mn of 4.7x103 and 14.0x103, it was found that the effect of molecular weight on the heat of crystallization ΔHc (0.3 J/g) was small, but the crystallization temperature increased by 12 °C, indicating that a higher kinetic energy was required for the decomposition of intermolecular bonds for long-chain PLA, while the free energy of crystallization of PLA remained constant. This reflects the limitation of PLA crystallization. However, the trend of the polymerization degree distribution (Mw/Mn) on the thermal properties of PLA is not obvious.
3.2 Thermal transformation and crystallization of PLA
L-lactic acid and D-lactic acid stereoisomers are naturally present as products of microbial activity. However, L-lactic acid is more common, and it also occasionally exists as a racemic mixture. Typical low molecular weight lactic acid L- and D-types can form racemic crystals when mixed together. This can be achieved in the cyclic form of lactic acid, called L-propyl cross-ester or D-propyl cross-ester, which has a melting point of 97.5°C, while its racemic compound melts at 124°C. The blend of PDLA and PLLA can form a three-dimensional composite structure with a melting point of 230°C, which is much higher than the 180°C of the respective pure PDLA and PLLA. This was demonstrated by Ikada et al. in a study using X-ray diffraction, showing the difference in crystal structure when PDLA and PLLA are co-blended to form a composite structure. Tsuji et al. cited the results of Sakakihara et al: When equimolar ratios of optically active polymers are mixed together, optical compensation occurs in the stereo polymers, leading to the formation of inactive materials in the crystal regions or units. The side-by-side stacking of stereo complexes is expected to form dense and ordered structures at higher melting temperatures.
PLLA is a semi-crystalline polymer with a melting point range of about 180°C and a crystallinity of about 70%. It can be processed by injection molding and extrusion processes. This L-shaped polymer appears to have the slowest degradation rate of all absorbable PLAs due to its high crystallinity. The semi-crystalline PLA has a higher shear viscosity than the amorphous PLA. However, the shear viscosity of both amorphous and semi-crystalline PLAs decreases with increasing temperature. Semi-crystalline PLA has both Tg and Tm. At temperatures above Tg (i.e. >58°C), PLA is rubbery, while below Tg it becomes glassy, but its molecules are still capable of creeping until it is cooled to its transition temperature below about -45°C, when it behaves as a brittle polymer. However, PDLLA is an amorphous polymer with no melting point and its Tg is between 50 and 60°C. Therefore, polymerization can be easily carried out in the melt, preferably in a reactor capable of handling highly viscous media. In contrast, the melting temperature of the stereocomposite PLLA-PDLA blend is 220-230°C, which is higher than that of PLLA and PDLA, and the PLLA-PDLA blend also has higher resistance to hydrolysis compared to pure PLLA and PDLA.
PLLA and PDLA are crystalline polymers, which is attributed to the enantiomeric purity of the original monomer and the steric regularity of the polymer chains. However, PDLLA, made by random copolymerization of equimolar amounts of L-propyleneglycerides and D-propyleneglycerides, usually remains in an amorphous form due to its irregular structure. However, most PLAs are crystalline because most lactic acids originate from L-isomers produced by microbial activity. auras et al. reported that PLAs can crystallize in three forms (a, β and γ) depending on the composition of the optically active L- and D,L-enantiomers. The α -structure is more stable with a melting point of 185 °C compared to the β -structure with a TM of 175 °C. The D and L isomers of PLA do not differ significantly in their thermal properties, and the D,L-propyl cross-esters exhibit amorphous behavior at the same molecular weight. In other words, the significant differences between the two D,L types and the D or L isomers lead to differences in the microstructure. Typical thermograms of L-PLA and D,L-PLA are shown in Figure 3-3 (Ahmed, Varshney, 2011). The peaks at 171.97 °C and 101.77 °C [Figure 3-3(a)] indicate the melting point and crystallization of PLLA, and PDLLA is an amorphous polymer that exhibits glass transition only at 52.73 °C [Figure 3-3(b)].
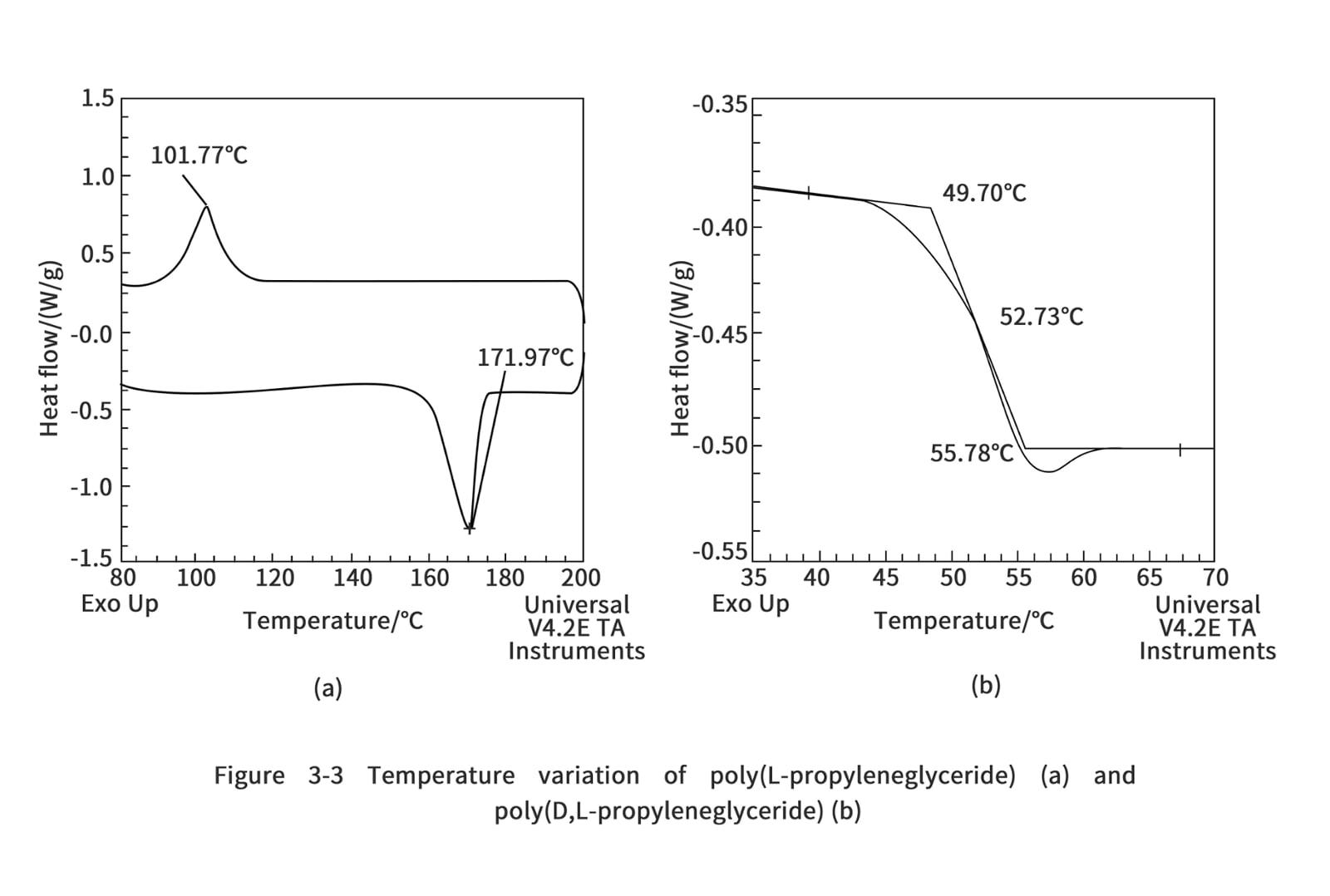
∞ Therefore, the Tg of PLA depends on the molecular weight and optical purity of the polymer. PLAs with higher L-propyleneglyceride content have higher Tg values than polymers with the same amount of D-propyleneglyceride, as reported by Dorgan et al. (Dorgan et al., 2005). In general, the relationship between Tg and molecular weight can be expressed by the Flory-Fox equation:
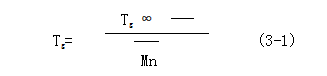
where Tg ∞ is Tg at infinite molecular weight:K is a constant indicating the excess free volume of the terminal group of the polymer chain; Mn is the average molecular weight. In the literature for PLLA and PDLLA, Tg∞ and K values were about 57-58°C and (5.5-7.3) × 104, respectively.
Lim et al. reported that thermal history has a significant effect on the glass transition behavior of PLA. The polymer melt is rapidly cooled or quenched (>500°C/min) to produce a highly amorphous polymer. This condition often occurs during injection molding and can lead to shrinkage, warping and even opacity of the molded product. Figure 3-4 show typical DSC analysis of two types of amorphous PLA, i.e. PDLLA (Mw = 70,000) samples that were originally amorphous and PLLA (M w = 200,000) samples that were quenched to amorphous form by rapid cooling at 100°C/min after melting. In both cases, Tg is evident at about 65 °C. However, the Tm of PLA is also related to its optical purity. The maximum practical Tm that can be obtained for pure chemically constructed PLA (L or D) is about 180°C, with an enthalpy of 40-50 J/g. However, the Tm values of PLA are generally in the range of 130 to 160 °C. The Tm suppression effect of endo-propyleneglycerides in polymerization can have a positive impact as it can improve processing properties by reducing thermal and hydrolytic degradation, or lead to reverse reaction of propyleneglyceride formation. The crystallization conditions affect the way PLLA crystallizes into three different crystalline forms, α, β and γ. X-ray diffraction results of PDLA and PLLA co-blended stereocomplexes show that the crystal structures are not the same as those of the homopolymers.
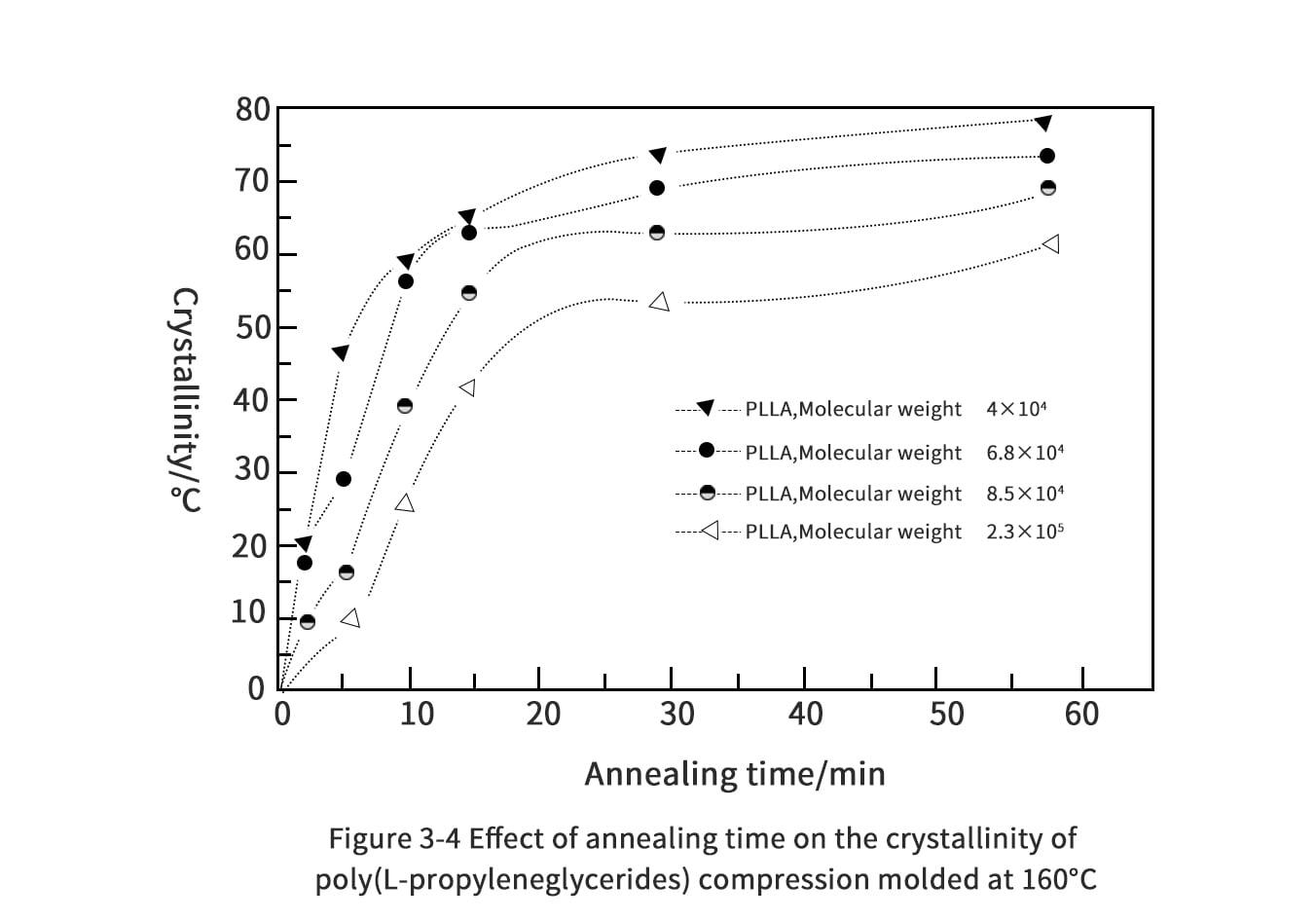
According to Auras et al, annealing between 75°C and the melting point of amorphous PLA can improve the crystallinity of PLA. This applies to the initially crystallizable PLA copolymer, i.e., the PLA should have good stereochemical purity. In addition, as shown in Figure 3-5, the crystallinity of the polymer increases with increasing annealing time and decreasing molecular weight (Mw), which is very favorable. Migliaresi et al. showed that slow annealing can gradually promote the movement of chain segment rearrangements in the crystal structure. Also, different cooling rates lead to changes in crystal morphology, with regular geometry and well-defined spherical crystals forming at high subcooling degrees and irregularly shaped and coarsely structured spherical crystals forming at lower decomposition temperatures.
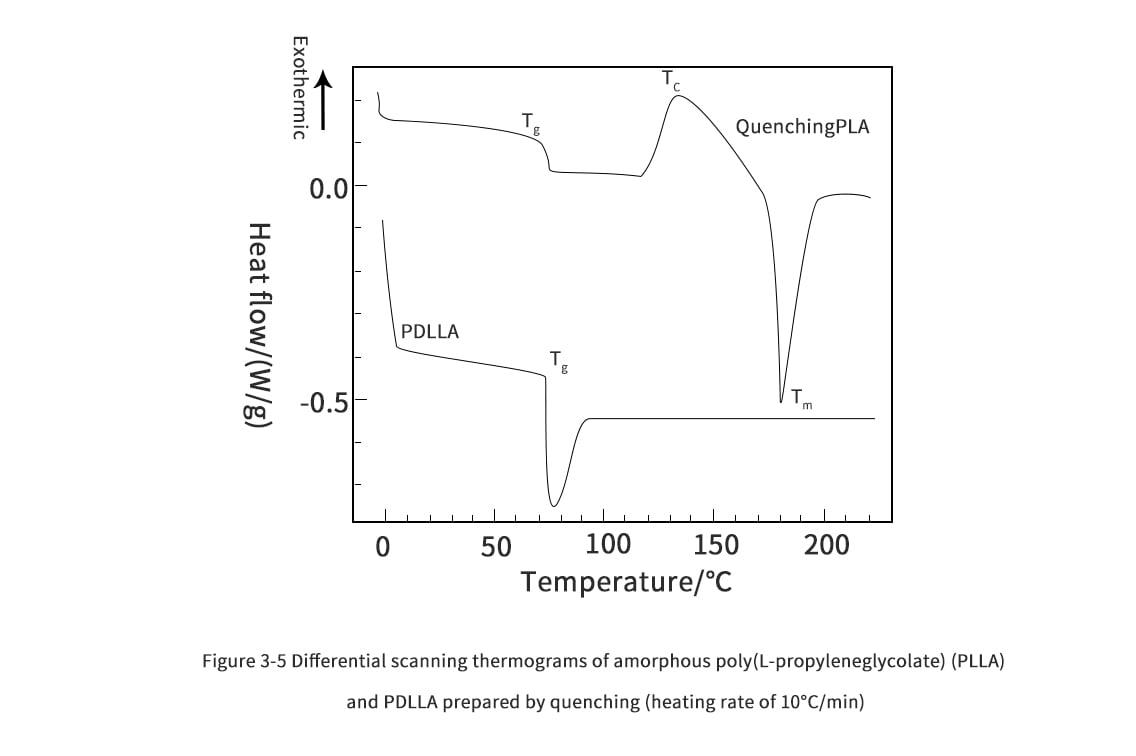
Typically, the addition of plasticizers can have a significant impact on the properties of the polymer. The addition of plasticizers can induce flexibility in rigid polymers and reduce Tm and Tg, thus significantly improving processability. Although an amorphous structure can be attracted by copolymerization of L-propyl cross-ester and D-propyl cross-ester isomers, thus reducing Tg, Kulinski and Piorkowska showed that the Tg of amorphous PLA is only 1 to 2 K lower than that of crystalline PLA [T(K)=t+273(°C)]. However, when only 5% of monomethyl ether polyethylene glycol was added as a plasticizer, the Tg of crystalline PLA decreased significantly from 59°C to 35-37°C, as shown in Figure 3-6. When the content of plasticizers increases to 10%, the Tg of all polymers drops sharply below room temperature and becomes almost indistinguishable. The plasticization of PLA by polyethylene glycol (PEG) and monomethyl ether of PEG was effective in reducing the Tg due to the presence of plasticizers which resulted in increased fluidity of PLA chain segments and with increasing plasticizer content. However, there was no evidence that the reaction of the monomethyl ether end groups affected the Tg of the crystalline PLA, as shown by comparing the curves of P550 (normal grade polyethylene glycol) and P600 (monomethyl ether polyethylene glycol). This can be attributed to the fact that the crystallinity of PLA has a stronger affinity for intermolecular interactions than the addition of plasticizers.
The role of propylene glycol as a PLA plasticizer is obvious, however, it tends to migrate to the surface of the material and make the surface sticky. In the long run, PLA products will become hard due to excessive plasticizer loss. Baiardo et al. compared the effects of monomeric plasticizers acetyl tributyl citrate (ATBC) and PEG on the thermal properties of PLA. They found that the compatibility limit of ATBC is 50% (mass fraction), while the compatibility of PEG in PLA decreases with increasing molecular weight. The compatibility of typical PEGs is 30% and 5% (mass fraction) at molecular weights Mw = 400 and Mw = 10000, respectively. In other words, plasticizers have better plasticizing effects at lower molecular weights. In recent years, other plasticizers such as glucosylglycerol and some fatty acid esters (Hoffman, 2002) have also been used to improve the flexibility and impact resistance of PLA.
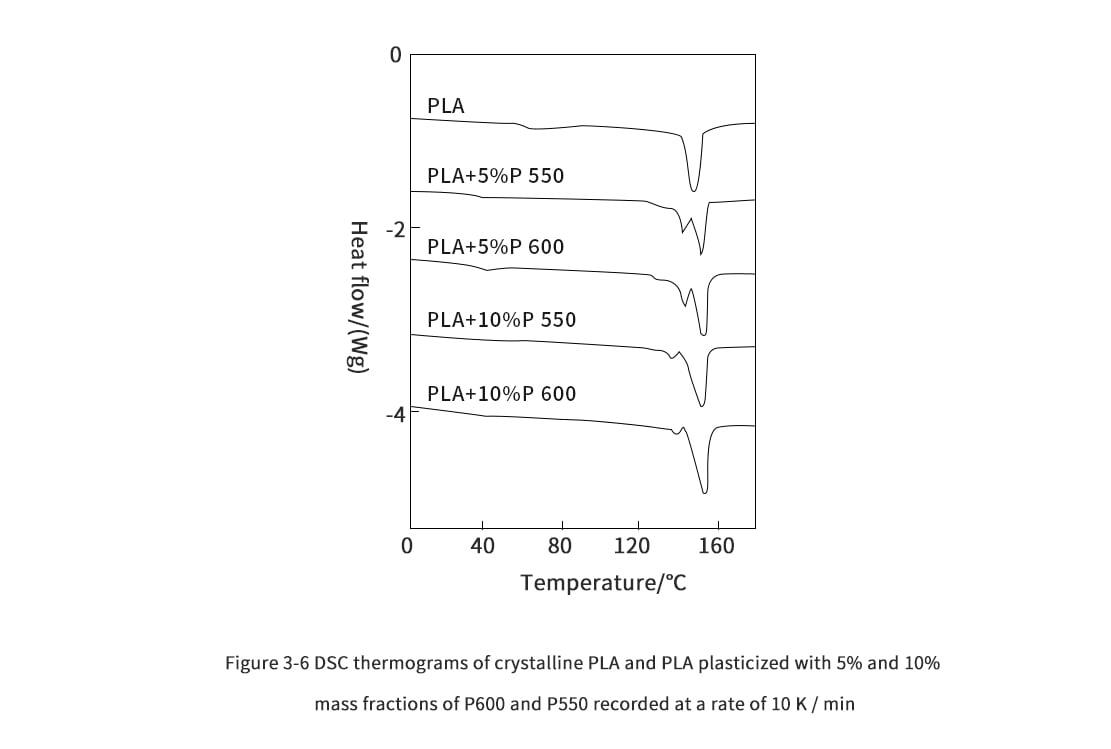
Pilin et al. extended their study on the effect of food grade plasticizers in PLA as shown in Table 3-2. The solubility parameter δ and the interaction parameter X were used to evaluate the compatibility of PLA with plasticizers. When the δ of the components is close to or X < 0.5, the mixture can be considered compatible and there is no phase separation. The DSC results shown in Table 3-3 indicate that for all plasticizers and components, the melt heat absorption of PLA is transferred to the low temperature fraction. However, the melting enthalpy (ΔHm) of low molecular weight PEG increases, which helps to explain the macroscopic phase separation phenomenon of PEG-200 and PEG-400. The ability of PBOH, acetylglycerol monolaurate and dibutyl sebacate (DBS) to induce PLA crystallization was rather weak, while PEG promoted crystallinity to values close to that of crystalline PLA (55 J/g), as shown in the results of Younes and Cohn. In addition, Pilin et al. reported that this phenomenon is due to the enhanced mobility of PLA macromolecules by PEG with higher mobility. Compatibility at the macromolecular scale is the ideal condition for obtaining significant PLA chain mobility. Martin and Avérous also found that the addition of various types of plasticizers, such as glycerol, polyethylene glycol, citrate, polyethylene glycol monolaurate (M-PEG), and oligomeric lactic acid, induced crystallization and promoted fusion in PLA (Table 3-4). It is believed that plasticizers can increase the chain mobility of laminar rearrangements and thus improve crystallinity.
Table 3-2 Solubility parameters δ and interoperability parameters χ between PLA and plasticizers
Name | Mw | δ(0.5MPa) | χ |
Polylactic Acid(PLA) | 74000 | 23.1 |
|
Poly(1,3-butanediol) (PBOH) | 2100 | 21.3 | 2.3 |
Dibutyl sebacate | 314 | 17.7 | 3.7 |
Acetylglycerol monolaurate(AGM) | 358 | 18.5 | 1.5 |
Polyethylene glycol(PEG-200) | 200 | 23.5 | 0 |
Polyethylene glycol(PEG-400) | 400 | 22.5 | 0.1 |
Polyethylene glycol(PEG-1000) | 1000 | 21.9 | 0.5 |
Table 3-3 Melting temperatures and enthalpies of pure components and PLA/plasticizer blends
| 100% | 100% | 10% | 10% | 20% | 20% | 30% | 30% |
Tm/℃ | ΔHm/(J/g) | Tm/℃ | ΔHm/(J/g) | Tm/℃ | ΔHm/(J/g) | Tm/℃ | ΔHm/(J/g) |
154.0 | 0.5 | — | — | — | — | — | — |
— | — | 148.0 | 34.1 | — | — | — | — |
6.9 | 113 | 150.8 | 32.4 | 142.4 | 44.6 | — | — |
39.8 | 149.4 | 153.0 | 32.1 | 150.6 | 38.6 | 149.3 | 41.3 |
—15.5 | 1.8 | 152.5 | 1.3 | 151.9 | 23.9 | 151.0 | 34.3 |
—8.3 | 71.9 | 150.3 | 1.6 | 146.6 | 29.3 | 143.4 | 31.4 |
—6.9 | 160.8 | 148.8 | 2.2 | 144.2 | 32.3 | 143.4 | 32.0 |
LIC's high-purity PLA is a semi-crystalline polymer with a Tg of 55°C and a Tm of 180°C. Changes in monomer type can significantly alter the structural properties of PLAs. For example, there are poly(L-lactide-co-D, L-lactide) copolymers with D, L-lactide content up to 30% (mole fraction) on the market, and Tg of 40-50℃, Contains up to 70% glycolide amorphous compound (D, L-lactide-co-glycolide) copolymer. The properties vary because the polymer is randomly distributed by the copolymer monomer. Due to the introduction of irregularities, the Tg of PLA copolymers is approximately proportional to the content of ethyl cross-ester or ε-lactone copolymer monomers. Furthermore, stereochemical defects in PLLA reduce the Tm, crystallization rate, and crystallinity of the resulting polymer to levels close to the properties of the copolymer monomer. In addition, the addition of fibers also causes changes in the thermal transition of PLA. A study by Gregorova et al. found that the addition of 20% (mass fraction) untreated natural fibers of North American spruce (also known as western Canada spruce, grown in North America) resulted in an increase in Tg to 52-54°C and an increase in crystallinity to 25.0%-28.7% with no change in Tm compared to pure PLA. The PLA used had a Tg of 46°C, a Tm of 150°C, and a crystallinity of 18.2%. This effect is due to the restricted mobility of the PLA chains in the presence of fibers. Jang et al. also observed that Tm was constant when PLA and starch were mixed (Table 3-5). The addition of starch resulted in little change in Tg and Tm, but a decrease in the heat of melting. Further studies using maleic anhydride as a compatibilizer showed that the Tg of PLA with natural blends was also reduced. Although maleic anhydride is introduced as a compatibilizer, it will have a plasticizing effect. This is because maleic anhydride does not produce a reinforcing effect, but increases the adhesion of natural fibers and PLA to obtain better elongation and avoid the formation of voids, thus avoiding premature failure during loading.
Table 3-4 Thermal properties of polylactic acid (PLA) and plasticizers
Materials | Tg/℃ | Tc/℃ | Tm/℃ | Crystallinity /% |
Pure PLA | 58 | — | 152 | 1 |
PLA-10%Glycerin | 54 | 114 | 142 | 24.3 |
PLA-20%Glycerin | 53 | 110 | 141 | 25.4 |
PLA-10%Citrate | 51 | — | 144 | 12 |
PLA-20%Citrate | 46 | — | 142 | 20 |
PLA-10%Polyethylene glycol monolaurate | 34 | 94 | 148 | 22 |
PLA-20%Polyethylene glycol monolaurate | 21 | 75 | 146 | 24 |
PLA-10%Polyethylene glycol | 30 | 82 | 147 | 26 |
PLA-20%Polyethylene glycol | 12 | 67 | 143 | 29 |
PLA-10%Oligolactic acid | 37 | 108 | 144 | 21 |
PLA-20%Oligolactic acid | 18 | 76 | 132 | 24 |
Table 3-5 Thermal properties and molecular weight of poly(lactic acid)-starch blends
PLA/Starch ratio /(Quality ratio)) | Maleic Anhydride /part | Tg/℃
| Tm/℃
| Crystallinity/% | Mn
| Mw
| Mw / Mw
|
100/0 | — | 63 | 154 | — | 95000 | 231000 | 1.6 |
90/10 | — | 62 | 154 | 2 | 49000 | 125000 | 2.5 |
80/20 | — | 61 | 153 | 2 | 39000 | 76000 | 1.9 |
70/30 | — | 61 | 153 | 9 | 33000 | 66000 | 2.0 |
60/40 | — | 62 | 154 | 10 | 44000 | 78000 | 1.8 |
50/50 | — | 62 | 153 | 11 | 42000 | 76000 | 1.8 |
90/10 | 3 | 61 | 153 | 12 | 41000 | 74000 | 1.8 |
80/20 | 3 | 57 | 154 | 18 | 41000 | 77000 | 1.9 |
70/30 | 3 | 59 | 155 | 36 | 47000 | 86000 | 1.8 |
60/40 | 3 | 60 | 155 | 48 | 45000 | 82000 | 1.8 |
50/50 | 3 | 60 | 155 | 41 | 44000 | 84000 | 1.9 |
In the study by Jang et al. the addition of starch increased the crystallinity of PLA blends. The crystallinity of PLA-starch blends was increased due to the starch-induced nucleation effect (Table 3-4). A comparison of the homogeneous and heavy homogeneous molecular weights (Mn and Mw) of different PLA-starch blends and pure PLA showed that the molecular weight was significantly reduced by almost half with the addition of only 10% starch. This significant decrease in molecular weight is thought to be due to the presence of starch moisture that triggers hydrolysis reactions in PLA. It should be noted that the crystallinity of the maleic anhydride (MA) capacitated blends is much higher than that of other blends with similar starch content. The crystallinity of MA capacitated blends increased with the increase of starch content. The capacitor also enhanced the regularity of the structural chain arrangement in the PLA-starch blends. When comparing the morphology of MA-enhanced PLA starch blends, the blended system forms a continuum without edges, holes, or cavities. This crystallinity does not affect the bonding strength; therefore, Tg and Tm remain constant due to the migration of MA into PLA during the blending process, although the heat of melt increases.
PLA is considered as a bio-based polymer that can replace petroleum-based polymers in the polymer industry. However, the poor thermal stability of PLA at temperatures below its melting temperature greatly limits the use of PLA in industry. The ester bonds of PLA degrade, so PLA tends to degrade during melt processing such as extrusion, injection molding processes and batch mixing. In addition, the low glass transition temperature Tg of PLA (about 60°C) also leads to its poor heat resistance. The effect of dwell time and temperature on the molar mass reduction of PLA for different melt processing methods has been studied by some. Since T-Tg has an effect on the activity of PLA chains, the glass transition temperature of PLA plays an important role in the crystallization of PLA substrates. According to Saeidlou et al, they concluded that the glass transition temperature increases sharply when the molecular weight of PLA increases from 80 to 100. However, as the molecular weight of PLA increases further, the glass transition temperature remains at a constant value. In addition, the chain structure of PLA also affects the glass transition temperature of PLA. PLA with a linear chain structure has a higher glass transition temperature value compared to PLA with a branched chain structure. This is because linear PLAs have a more compact structure than branched PLA chains with a higher number of terminal groups. Pitet et al. found that the glass transition temperature of branched PLAs is lower than that of linear PLAs with the same molecular weight. This is due to the presence of higher free volume in the matrix of branched PLA polymers and the fact that PLA molecular chains have more terminal groups. In addition, Zhao et al. also found that their prepared star-shaped PLA with 32 arms had a lower glass transition temperature than linear PLA. Le Marec et al. tested unprocessed PLA resin and determined its glass transition temperature and melting temperature to be 58 °C and 150.8 °C, respectively, while no crystallization peak appeared on the DSC curve. This is because the PLA resin has crystallized during the manufacturing process.Le Marec et al. found that the processing conditions of the original PLA did not have a significant effect on the glass transition temperature (~56°C). This is due to the fact that the processing conditions did not change the chain structure of PLA.
3.2.1 Effect of crystallization temperature on crystal shape
The formation of different types of crystal structures in PLA substrates is basically influenced by the crystallization process conditions. Studies by De Santis and Ko- vacs and Kalb and Penning found the presence of α-type crystals in the matrix of PLA polymers treated by conventional melt and solution crystallization. They also suggested that α-type crystals are formed by the 103-helix chain conformation during melt crystallization and cold crystallization. A study conducted by Zhang et al. found that when PLA crystallizes below 120 °C, a different type of crystal, α' type crystal, is formed in the PLA matrix. The structural system of α' type crystals is similar to the structure of α-type crystals, but the chain stacking arrangement in α' type crystals is looser and more randomly arranged than that of α-type crystals. However, Zhang et al. and Kawai et al. further pointed out that α' type crystals are formed only at crystallization temperatures below 100 °C. They also indicated that the α-type crystal structure and α' type crystal structure coexist in the PLA matrix when PLA is crystallized at 100-120°C. Compared with the α-type crystal structure, the α' crystal structure has loose chain stacking and less ordered structure, which makes its modulus low and its barrier property poor. Eling et al. showed that the α-type crystal structure was partially changed and transformed into β-type crystal when PLLA fibers were obtained by melt spinning or solution spinning process at high stretching temperature and high stretching ratio, and that Hoogsteen et al. found that the α-type crystal structure has a laminar folded chain structure morphology by small angle X-ray scattering experiments. In addition, their results also indicate that the β-type crystal structure is caused by the deformation of the laminar folded chain structure, which corresponds to the fibrous filament crystal. In addition, Hoogsteen et al. obtained a melting temperature of about 185°C for α-type crystal structures and about 175°C for β-type crystal structures. This is due to the chain arrangement structure in β-type crystals forming a less than perfect chain stacking structure under high tensile stress. This is the main reason why α-type crystal structures with well-layered folded chain structures are more stable than β-type crystal structures. Cartier et al. reported γ-type crystal structures formed by epitaxially crystallized PLA on the crystal structure of hexamethylbenzene, in which two chains are arranged antiparallel and formed in a single unit cell.
In general, PLA substrates are known to have three different types of phenotypes, namely α-, β- and γ-type crystal structures. these crystal fractions of PLA are influenced by the crystallization temperature Tc. Zhang et al. analyzed the effect of crystallization temperature on the behavior of PLA by performing DSC analysis in the temperature range of 85-150 °C. They studied the thermal behavior of PLA with three different molecular weights (980000, 150000 and 50000) when the crystallization temperature was increased from 85°C to 150°C. The selected crystallization temperature range is higher than the glass transition temperature of PLA (Tg ≈ 58°C). They found three different melting behaviors of PLA samples with molecular weight of 150,000 at crystallization temperatures ranging from 85°C to 150°C. The appearance of melting peaks (Tm1 and Tm2) is mainly attributed to the melting behavior of α-type crystal structures in the PLA matrix. This can be further demonstrated by the XRD observations of Zhang et al. where the crystal deflection peak 203 shifted to higher 2θ values indicating that the PLLA samples crystallized in this temperature range to form α' and α' type crystal structures, as shown in Figure 3-7. A single melt peak Tm2 was observed on the DSC curve of PLA when the crystallization temperature was above 130 °C, as shown in Figure 3-7, indicating that the melt peaks of Tm1, and Tm2, were combined into one melting point. This is because as the crystallization temperature increases, there is greater densification of the stacked chains in the crystal, resulting in smaller lattice spacing in the crystal. Therefore, at higher crystallization temperatures, the α' type crystal structure is completely transformed into stable α-type crystals.
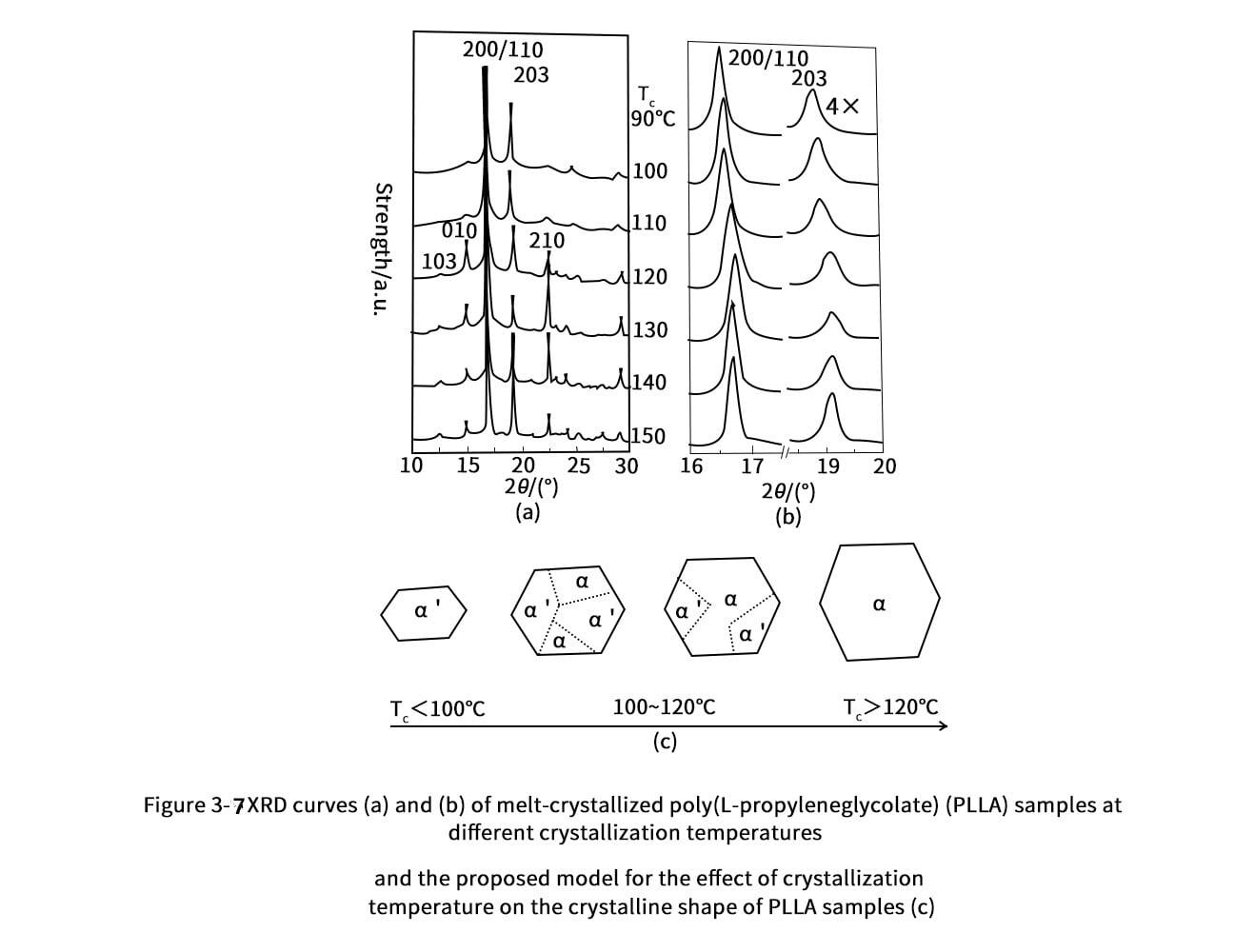
A study conducted by Le Marec et al. found that the crystallization temperature of PLA treated at a constant temperature of 170°C for 10 min decreased gradually as the rotational speed was increased from 40 to 150 r/min. In addition, the molecular weight of PLA decreased gradually with the increase of processing speed. When the processing time was increased to 30 min and the rotational speed was increased from 40 r/min to 75 r/min, the crystallization temperature of PLA was increased from 104.5°C to 106.6°C. However, further increasing the speed from 75r/min to 150r/min will reduce the crystallization temperature. The study conducted by Le Marec et al. also showed that the reduction of PLA molecular weight during processing can promote PLA crystallization by lowering the crystallization temperature. This is attributed to the fact that the reduction in PLA molecular weight leads to a better molecular mobility of PLA chains, which can crystallize into a more ordered arrangement. On the other hand, Liuyan et al. also investigated the effect of crystallization temperature on the crystallization rate of PLA. They found that increasing the crystallization temperature significantly delays the crystallization rate because higher temperatures affect the mobility of PLA molecular chains and lead to the formation of unstable nuclei, thus requiring more time to complete the crystallization process.
3.2.2 Effect of nucleating agents on PLA crystallization
PLA has good biocompatibility and biodegradability and has received a lot of attention from the polymer engineering community in recent years. PLA is a biodegradable polymer that can be buried in soil and decomposed into water and carbon dioxide. Due to its biocompatibility and biodegradability, PLA can replace petroleum-based polymers and is widely used in medical devices, food packaging, tableware, etc. However, the poor thermal stability, low mechanical properties such as hardness and brittleness, and slow crystallization rate of PLA limit its application in industry. In addition, these PLA products are typically produced using fast-cooling extrusion and injection molding processes, resulting in amorphous PLA products with low glass transition temperatures. In their study, Gui et al. investigated the effect of using various types of commercial nucleating agents (e.g. Millad 3988, Hyperform HPN-68L, WBG-II, TMB-5, Bruggolen P22, Bruggolen, P25, PET-C and TMC-328) to overcome these drawbacks. , PET- C and TMC-328). They found that the crystallinity of PLA increased from 0 to more than 35% with the addition of 0.5% (mass fraction) of PET-C and TMC-328, an organically modified montmorillonite clay, and TMC-328, an amide compound. Different researchers, including Gui et al, Feng et al, and Madera Santana et al, have studied that the original PLA shows a broad and small deflection peak 200/110 at 2θ≈16.5°. Gui et al. found that the addition of PET-C and TMC-328 greatly induced crystallization and sharpened the deflection peak 200/110, indicating an increase in grain size and crystallinity. In a study conducted by Feng et al. they investigated the nucleation effect of nucleating agents such as boric acid, potassium 3,5-bis(methoxycarbonyl)benzenesulfonate (LAK-301), substituted dibenzoyl phosphate (TMP-5), N′1,N′6-dibenzoyl dihydrazide (TMC-306), talc and N′1,N′6-(ethane-1,2-diyl)bis(N2-benzoxalamide) (OXA) Nucleation effects on the crystallization behavior of PLA. They found that the nucleation efficiency of TMC-306 and OXA nucleating agents was as high as 49%. In addition, they found that TMC-306 and OXA nucleating agents had a very significant effect on reducing the crystallization time of PLA. Wang et al. investigated the effect of increasing the addition of nucleating agent TMC-328 from 0.2% (mass fraction) to 0.6% on the crystallization behavior of PLA. They found that the crystallinity of PLA gradually decreased with increasing the amount of TMC-328. The results showed that increasing the amount of nucleating agent TMC-328 could improve the crystalline properties of PLA composites. In addition, Xu et al. conducted a similar study on the effect of the addition of the hydrazide complex nucleating agent TMC-306 on the crystallization behavior of PLA. Increasing the addition of TMC-306 from 0.05% (mass fraction) to 0.5% significantly increased the height of the deflection peak 200/110 on the XRD curves of PLA blends and sharpened the peaks. They found two distinct peaks at 2θ ≈ 16.7° and 19.1° on the XRD curve, which is an α-type crystal structure of the original PLA. They also found that the addition of TMC-306 accelerated the isothermal crystallization process of PLA as a whole. In conclusion, the addition of nucleating agent can induce the crystallization behavior of PLA by accelerating the crystallization rate.
3.3 Thermal decomposition
The pyrolysis of PLA depends on a number of factors such as molecular weight, crystallinity, purity, temperature, pH, presence of terminal carboxyl or hydroxyl groups, water permeability and catalytic additives, which may include enzymes, bacteria or inorganic fillers. Celli and Scandola and Sodegard and Stold showed that PLLA is sensitive to thermal decomposition and that the thermal decomposition of PLA is influenced by the following factors:
(1) Hydrolysis of trace amounts of water, catalyzed by the hydrolysis monomer, lactic acid (Figure 3-8);
(2) The chain-like depolymerization was catalyzed by the remaining polymerization catalyst (Figure 3-9);
(3) Oxidation, random main chain breakage ;
(4) The intermolecular ester exchange reaction produces monomers and oligoesters (Figure 3-10) or non-intermolecular ester exchange reactions produce low molecular weight monomers and oligolactic acid.
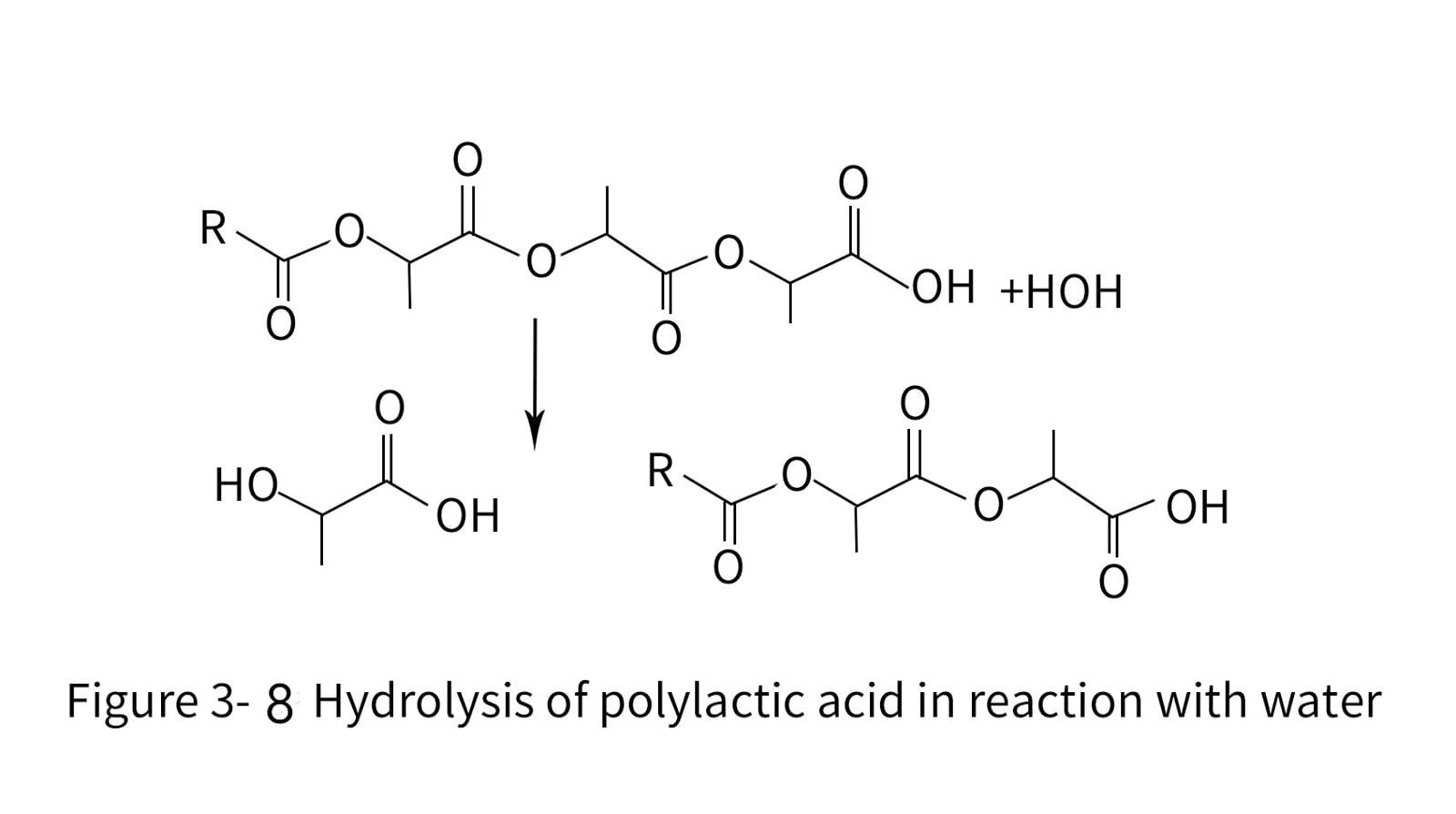
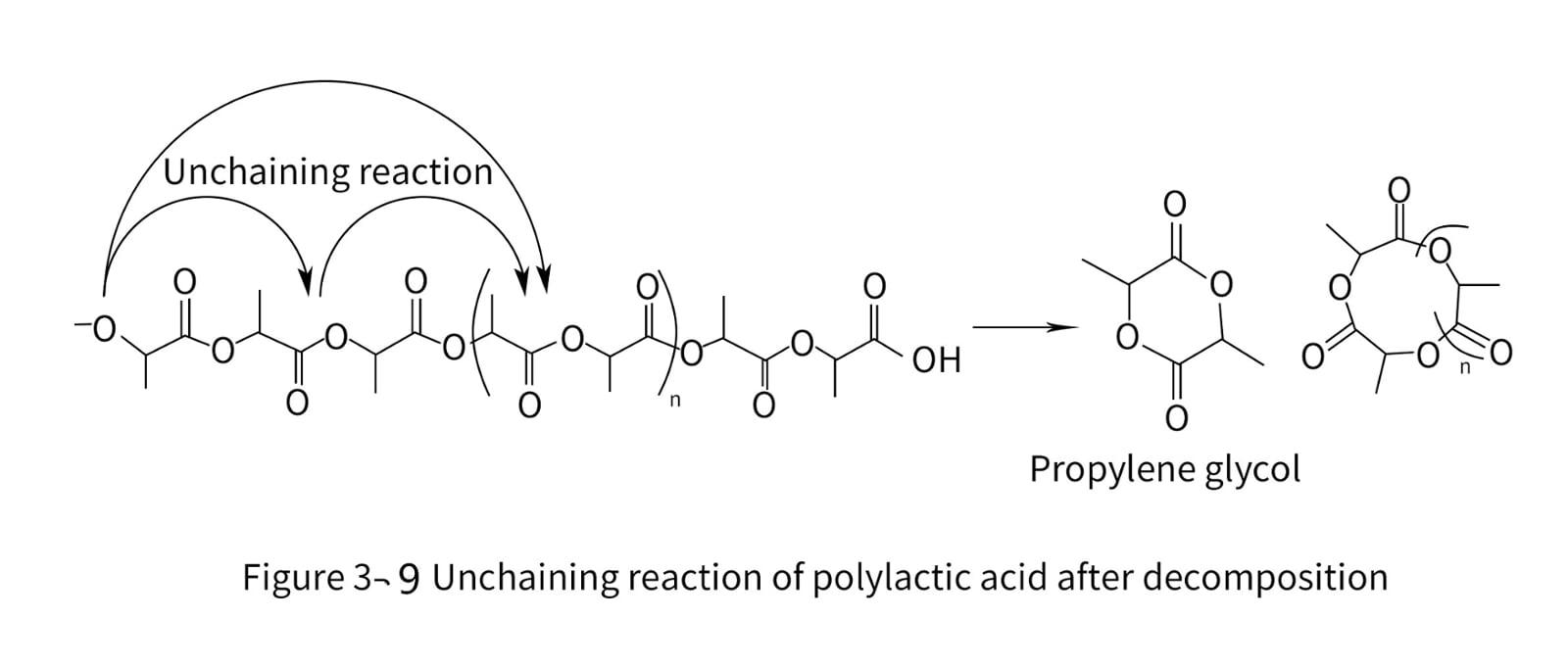
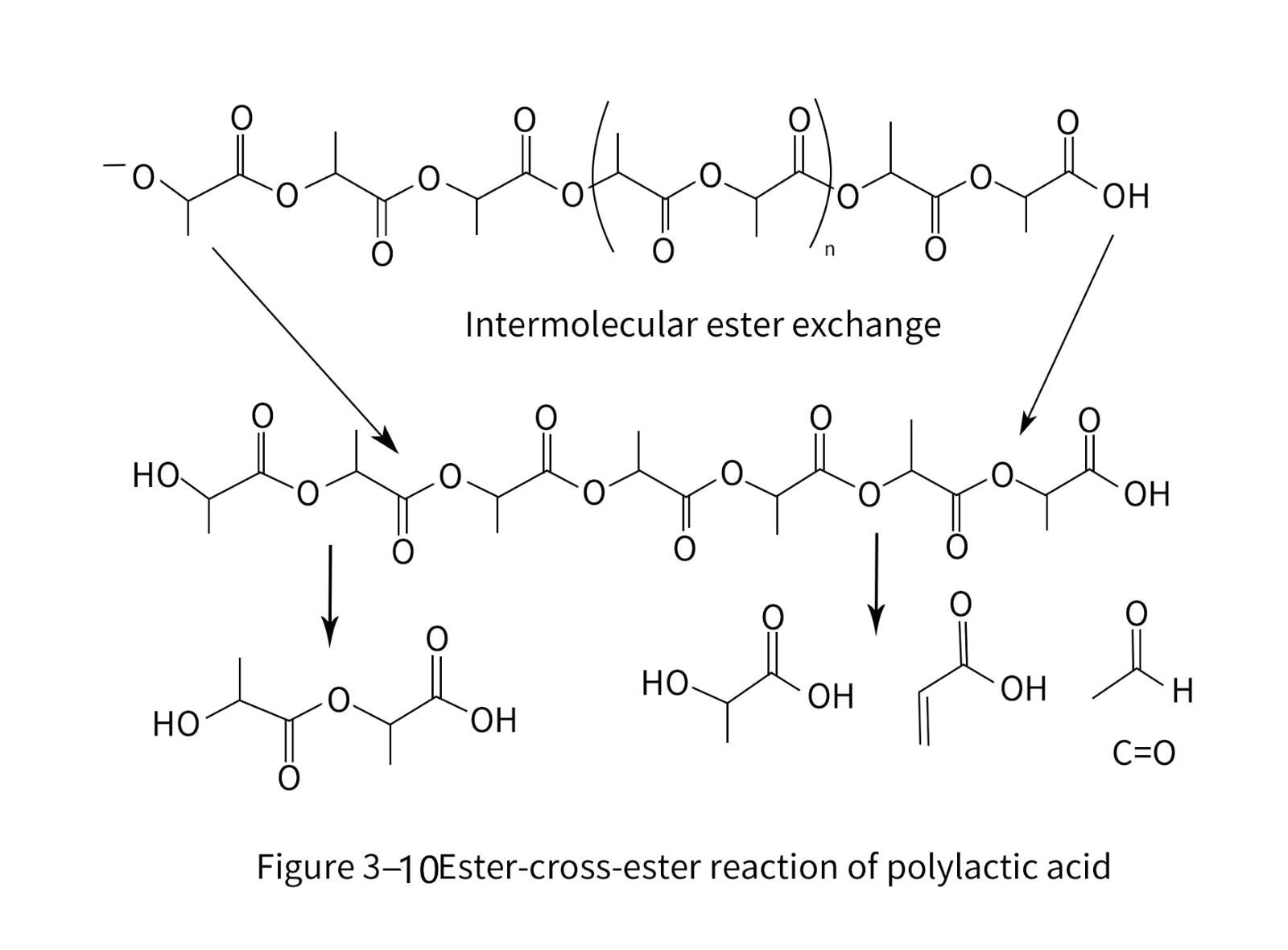
The decomposition temperature of PLA is generally 230-260°C. Therefore, it is safe to use PLA at room temperature. PLA is rarely used at high temperatures, such as for boiling water, because it loses its structural properties at temperatures greater than 60°C. Although PLA is unlikely to release significant amounts of toxic substances, further attention needs to be paid to residual plasticizers or oligomers. PLA undergoes initial thermal decomposition at temperatures above 200°C, followed by hydrolysis reactions, then propyleneglyceride recombination, oxidative backbone breakage and intermolecular ester exchange reactions. Thermal decomposition can occur at 200 °C in the absence of a catalyst, but higher temperatures are required to induce faster and more extensive reactions.
Since PLA is one of the polymers that are highly sensitive to heating, many researchers have conducted studies to modify PLA. McNeil and Leiper performed isothermal degradation of PLA at several temperatures and found that the activation energy was 119 kJ/mol and the thermal degradation mechanism was considered to be initiated by segmental terminal esters in the temperature range of 240-270 °C. Chain cleavage occurs to produce cyclic oligomers, propyl cross-ester, acetic acid and carbon monoxide, and finally carbon dioxide and methyl ketone at higher temperatures. In a further study by McNeil and Leiper, oligomers were observed to account for more than 50% of the total volatile composition when the set heating temperature reached 440°C, the temperature at which volatilization is complete. Carbon dioxide, acetaldehyde, vinyl ketone and carbon monoxide were also formed in this volatile gas stream. During the thermal decomposition, the acetylation reaction at the end of the chains increased the thermal properties of the polymer by about 30°C, suggesting the involvement of hydroxyl groups in the degradation of PLA. In addition, McNeil and Leiper added polymethyl methacrylate as a source of free radicals during the thermal decomposition of PLA. The decomposition rate of PLA increased while PMMA was stable, i.e., the yields of CO2 and PLA oligomers increased significantly, indicating that the free radical reaction is one of the important pathways for the formation of PLA oligomers at high temperatures. PLA tends to follow the dominant reaction pathway of intramolecular ester exchange of pure PLA to form cyclic oligomers, usually accompanied by residual carbon oxides and acetaldehyde from the cleavage reaction. However, when the PLA samples were contaminated with residual stannous catalyst, i.e. residual polymerization catalyst, the PLA underwent a selective depolymerization step, yielding only propyl cross-ester. This observation was further confirmed by Cam and Marucci, whose results showed that the residual metals have a strong thermal degradation effect on PLA. The degradation activity of metal residues follows the order of Sn<Zn<Al<Fe for the family of metals commonly used for PLA polymerization, i.e., Sn, Zn, Al, and Fe.
The depolymerization of PLLA at elevated temperatures leads to intra- and interester chain transfer and depolymerization reactions through significant changes in the number of specific spins. In other words, the high capacity of transition metals can coordinate the ester groups and accelerate the reaction.
3.4 Heat capacity, thermal conductivity and pressure-volume-temperature relationship of PLA
Heat capacity, thermal conductivity and pressure volume-temperature (PVT) are macroscopic properties of polymers that are important during processing. The heat capacity determines the amount of heat required to raise the volume of PLA to the final processing temperature. Meanwhile, thermal conductivity and PVT have an effect on the heat transfer rate and compressibility of injection molded products, which is important for determining the shrinkage of injection molded products.
Heat capacity is the amount of heat required to change the temperature of a substance by a certain value. It is important to determine the magnitude of the initial energy required to raise the temperature of the polymer to the processing temperature. A comprehensive study of PLA heat capacity was reported by Pyda et al. As shown by Pyda et al. the data shown in Table 3-6 are the most comprehensive to date and include a temperature range of 5 to 600 K.
Table 3-6 Measured, theoretical and recommended heat capacities of polylactic acid (PLA)
T/K | Cp(exp) is determined by the adiabatic heat method① /[J/K·mol﹚] | Cp(exp)② /[J/K·mol﹚] | Cp(Vibration)③ /[J/K·mol﹚] | Recommended experimental heat capacity④/[J/K·mol﹚] |
5 | 0.31 | NA | 0.46 | (Solid)0.31 |
6 | 0.60 | NA | 0.77 | 0.60 |
7 | 0.95 | NA | 1.17 | 0.95 |
8 | 1.34 | NA | 1.62 | 1.34 |
9 | 1.78 | NA | 2.11 | 1.78 |
10 | 2.25 | NA | 2.63 | 2.25 |
15 | 4.85 | NA | 5.30 | 4.85 |
20 | 7.74 | NA | 7.83 | 7.74 |
25 | 10.585 | NA | 10.22 | 10.585 |
30 | 13.15 | NA | 12.535 | 13.15 |
40 | 18.06 | NA | 17.04 | 18.06 |
50 | 22.585 | NA | 21.48 | 22.585 |
60 | 26.575 | NA | 25.86 | 26.575 |
70 | 30.455 | NA | 30.14 | 30.455 |
80 | 34.195 | NA | 34.27 | 34.195 |
90 | 37.77 | NA | 38.21 | 37.77 |
100 | 41.145 | NA | 41.195 | 41.145 |
110 | 44.40 | NA | 45.40 | 44.40 |
120 | 47.52 | NA | 48.66 | 47.52 |
130 | 50.52 | NA | 51.73 | 50.52 |
140 | 53.41 | NA | 54.64 | 53.41 |
150 | 56.2 | NA | 57.40 | 56.2 |
160 | 58.98 | NA | 60.50 | 58.98 |
170 | 61.71 | NA | 62.61 | 61.71 |
180 | 64.40 | NA | 65.10 | 61.40 |
190 | 67.08 | 69.76 | 67.54 | 67.08 |
200 | 69.75 | 68.23 | 69.95 | 69.75 |
210 | 72.43 | 72.27 | 72.34 | 72.35 |
220 | 75.13 | 74.78 | 74.71 | 74.96 |
230 | 77.87 | 77.14 | 77.08 | 77.51 |
240 | 80.65 | 79.61 | 79.44 | 80.13 |
250 | 83.50 | 82.10 | 81.81 | 82.80 |
260 | NA | 84.25 | 84.19 | 84.25 |
270 | NA | 87.11 | 86.57 | 87.11 |
280 | NA | 89.82 | 88.98 | 89.82 |
290 | NA | 92.48 | 91.35 | 92.48 |
298.15 | NA | 94.69 | 93.31 | 94.69 |
300 | NA | 95.30 | 93.75 | 95.30 |
Continued Table
T/K
| Cp(exp) is determined by the adiabatic heat method① /[J/K·mol﹚] | Cp(exp)② /[J/K·mol﹚] | Cp(Vibration)③ /[J/K·mol﹚] | Recommended experimental heat capacity④ /[J/K·mol﹚] |
310 | NA | 98.13 | 96.15 | 98.12 |
320 | NA | 101.59 | 98.55 | 101.59 |
330 | NA | 112.16 | 100.95 | 112.15 |
332.5(Tg) | NA | 123.57 | 101.67 | 145.44 |
340 | NA | 144.36 | 103.34 | (liquid)146.01 |
350 | NA | 144.40 | 105.74 | 146.77 |
360 | NA | 147.59 | 108.12 | 147.53 |
370 | NA | 148.24 | 110.49 | 148.29 |
380 | NA | 148.61 | 112.86 | 149.05 |
390 | NA | 149.91 | 115.21 | 149.81 |
400 | NA | 150.56 | 117.55 | 150.57 |
410 | NA | 151.62 | 119.87 | 151.33 |
420 | NA | 152.31 | 122.18 | 152.09 |
430 | NA | 152.97 | 124.47 | 152.85 |
440 | NA | 153.725 | 126.74 | 153.61 |
450 | NA | 154.29 | 129.00 | 154.37 |
460 | NA | 154.98 | 131.24 | 155.13 |
470 | NA | 155.77 | 133.46 | 155.89 |
480(Tm) | NA | NA | 135.66 | 156.65 |
490 | NA | NA | 137.84 | 157.41 |
500 | NA | NA | 140.01 | 158.17 |
510 | NA | NA | 142.15 | 158.93 |
520 | NA | NA | 144.28 | 159.69 |
530 | NA | NA | 146.40 | 160.45 |
540 | NA | NA | 148.49 | 161.21 |
550 | NA | NA | 150.57 | 161.97 |
560 | NA | NA | 152.64 | 162.73 |
570 | NA | NA | 154.69 | 163.49 |
580 | NA | NA | 156.72 | 164.25 |
590 | NA | NA | 158.74 | 165.01 |
600 | NA | NA | 160.75 | 165.77 |
① Experimental data on heat capacity according to the adiabatic heat method(Kulagina et al.,1982);
② The experimental data represent the average of three measurements performed on two samples, each using standard differential scanning calorimetry (DSC) and temperature modulation for 1.5% D isomer, 8.1% D isomer and 16.4% D isomer PLA;
③ The heat capacity of the solid PLA was calculated assuming only vibrational motion. For more information, see Pyda et al.(2004)。
④ Recommended experimental heat capacities for PLA in solid and liquid form。
The thermal conductivity of PLA increases almost with the increase of temperature. The thermal conductivity of the polymer has a great influence on the heat dissipation during the cooling process of the injection molded product. Adequate and controlled heat dissipation can reduce the possibility of warpage.
The PVT relationship of the polymer determines the compressibility of the molten polymer and the shrinkage of the finished product. This is especially important for those thick-walled products with complex designs (>5mm). When the molten polymer is cooled, it has been found that rapid cooling leads to the formation of amorphous structures. In other words, even though the polymer itself is crystallizable, the macromolecules of the polymer cannot form a crystalline structure. The specific volume of crystalline plastics undergoes a significant change. This is due to the fact that the crystal structure is highly dense. When a semi-crystalline polymer is injection molded in a specific closed channel, the resulting product often has some dimensional deviations, indicating shrinkage. Irregular shrinkage of hot products can also lead to warpage. Since PLA is a semi-crystalline polymer, heat treatment of PLA helps to eliminate shrinkage. During the injection molding process, high pressure helps compress the PLA molten polymer during the filling phase to produce a highly dimensionally stable product. A typical PVT can be modeled using the two-domain Tait-PVT model which is widely used in injection molding simulation software to predict problem areas in injection molded products.
The detailed explanation of the two-domain Tait PVT equation is as follows:
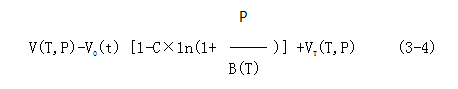
where V(T, P) - the specific volume at temperature T and pressure P;
V0 - Specific volume at zero gauge pressure;
T - temperature in K;
P - pressure in Pa;
C - a constant 0.0894.
The higher temperature region (T>Tt) can be described by the following equation:
V0=b1m+b2m(T-b5) (3-5)
B(T)=b3mexp [-b4m(T-b5)] (3-6)
Vt(T,P)=0
where b1m, b2m, b3m, b4m and b5 are the data fit coefficients.
The lower temperature region (T<Tt) can be described by the following equation:
V0-b1s+b2s(T-b5) (3-7)
B(T)=b3sexp[-b4s(T-b5)] (3-8)
Vt(T,P)=b7exp[b8(T-b5)-b9P] (3-9
where b1s, b2s, b3s, b4s, b5, b7, b8, and b9 are the data fit coefficients.
The dependence of Tt on pressure can be described by the following equation:
Tt(P)=b5+b6P (3-10)
where b5 and b6 are the data fit coefficients.
3.5 Conclusion
Thermal properties have a significant impact on the performance of PLA. The thermal properties of PLA are closely related to the crystallinity. Importantly, the L and D structure chemistry has an effect on the crystallization and thus on the melting temperature and glass transition temperature of PLA. Copolymers and additives contribute to improved heat transfer and better processing properties. At temperatures greater than 200°C, PLA undergoes severe degradation, including propylene glycol and oxide gas generation. Information on heat capacity, thermal conductivity and PVT is important for designing high quality marketable PLAs.
4. Chemical properties of polylactic acid
4.1 Introduction
Polylactic acid (PLA) has good biocompatibility and biodegradability, and can be easily decomposed by hydrolysis reaction. PLA is derived from renewable agricultural resources, such as maize and cassava, and the large-scale production of PLA can facilitate the consumption of agricultural products and contribute to the development of the agricultural economy. In addition, the replacement of traditional stone-based plastics with PLA can help reduce CO2 emissions due to the good carbon sequestration effect of agricultural activities.
PLA is a biodegradable polymer material that has been extensively studied and used in household packaging and biomedical applications such as absorbable sutures, surgical implants, tissue engineering scaffolds, and drug-controlled release devices. The monomeric lactic acid of PLA can be in two stereoisomers, called D and L types, or form a racemic mixture, called D,L type. D- and L-types are optically active, while D,L-types are optically inactive. Poly(L-lactic acid) (PLLA) and poly(D-lactic (PDLA) are semicrystalline, while poly(D,L-lactic acid) (PDLLA) is amorphous (Jain, 2000; Urayama et al., 2003).
PLA belongs to a family of aliphatic polyesters, usually made from alpha-hydroxy acids, and also includes polyglycolic acid, polyethylene lactone and polydioxanone. It is one of the few polymers with a stereochemical structure that can be easily modified by polymerizing controlled mixtures of L and D isomers to obtain high molecular weight amorphous or semi-crystalline polymers. The properties of PLA can be improved by changing the isomer (L/D) of PLA and copolymerizing with monomers such as ethyl cross ester and caprolactone. PLAs can also be formulated with the addition of plasticizers, other biopolymers and fillers. the biodegradability of PLA blends means that they are well suited for short-term packaging materials and also further extends the use of PLAs in biomedical applications where biocompatible properties are essential, such as implants, sutures and drug encapsulation.
In the early days of PLA development, PLA was produced by polycondensation (Figure 4-1). This is the most straightforward method for synthesizing PLA, but has the disadvantage of producing too much water and low molecular weight (Mn <1000 to 5000) products. Sometimes, chain extenders may be needed to increase the molecular weight, but this raises the cost of production. PLA can also be produced by azeotropic dehydration condensation. This polymerization technique yields high molecular weight polymers, but requires different diacids, diols or hydroxy acids and advanced catalysts (Garlotta). These components are left in the PLA as impurities and may produce unwanted degradation during subsequent high temperature processing.
The most important method for high-volume production of high-molecular-weight PLA is ring-opening polymerization. High molecular weight PLA is generated from cyclopentanedioic acid esters (often called propylenes), often with stannous octanoate as the catalyst. This mechanism does not produce additional water and therefore higher molecular weights can be obtained. The polymerization of racemic mixtures of L-propyleneglycerides and D-propyleneglycerides usually results in the synthesis of amorphous PDLLA. The application of stereotactic catalysts tends to produce stereochemically pure PLA with good crystallinity. The crystallinity and mechanical properties depend on the ratio of D/L enantiomers, which is also partially related to the type of catalyst used. High quality production of PLA has been reported to minimize the amount of unreacted lactic acid monomers and reduce lactic acid leaching when PLA is used as a packaging material. In addition, the amount of lactic acid exuded is much lower compared to the amount of lactic acid in common food ingredients (Mutsuga et al., 2008). Therefore, polymers made from lactic acid are excellent candidates for packaging applications (Iwata, Doi, 1998). PLA has been growing as an alternative packaging material in niche markets. PLA is currently used as a food packaging polymer for "short shelf-life" consumer products, including containers, mugs, razors and stationery, and PLA fibers are also used in carpets, sportswear and diapers. In recent years, new applications have been developed, such as electronic equipment housings and flooring materials, and PLA's "green" certification means that plastic materials have a sustainable future worldwide.
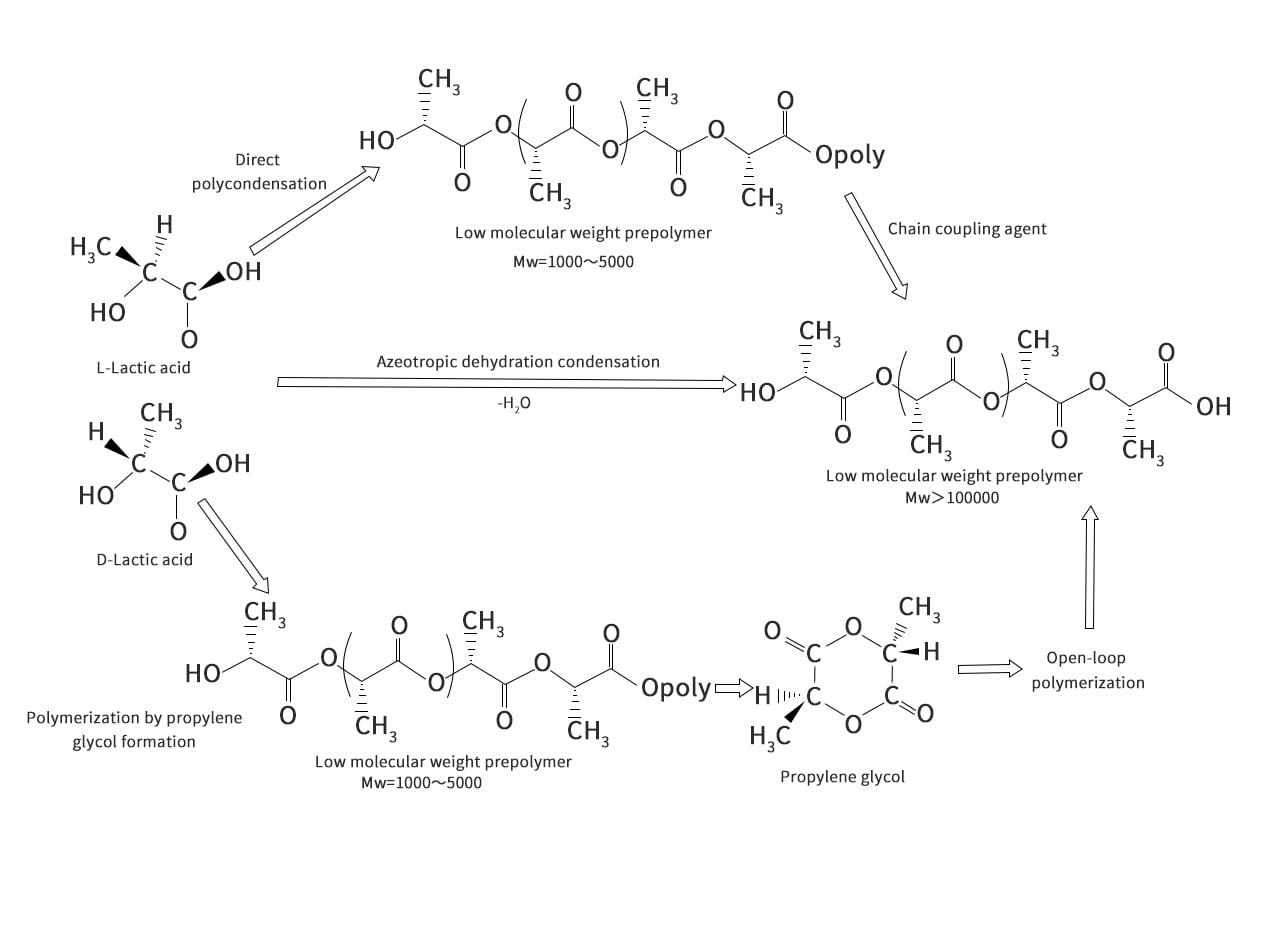
4.2 Establishment chemistry of polylactic acid
The basic component of PLA is lactic acid, which is produced by bacterial fermentation or petrochemical feedstock. Lactic acid is a naturally occurring substance whose standard chemical name is 2-hydroxypropionic acid. It is the simplest hydroxy acid with an asymmetric carbon atom and has optically active L(+) and D(i) isomers. Both L and D isomers are produced in bacterial systems, with the L-isomer being more common. Meanwhile, the mammalian system produces only the L-isomer, which is readily absorbed by the enzyme protease K. Figure 4-2 shows the chemical structures of L-lactate and D-lactate.

Lactic acid is now produced in large quantities by bacterial fermentation of carbohydrates, with maize and cassava being the main agricultural sources. There are about 20 genera in the thick-walled phylum of lactic acid producing bacteria (Reddy et al). Strains of Lactobacillus deuterium, Lactobacillus yanteri and Lactobacillus acidophilus produce D-lactic acid and some strains also produce mixtures (Nampoothiri et al.). Many fermentation processes now use a lactic acid bacterium with a high lactic acid yield.These bacteria can actively produce lactic acid and survive at low oxygen concentrations over a wide processing window, including a pH of 5.4 to 6.4 and a temperature of 38 to 42°C. The usual sources of monosaccharides in agriculture are widely used in lactic acid fermentation, such as glucose and maltose in corn or potatoes, sucrose in sugar cane or sugar beets, and lactose in cheese whey. Other nutrients, such as vitamin B complexes, amino acids and nucleotides, are needed throughout the process to ensure bacterial function; this nutrient pool provides a nutritious corn pulp.
Propyleneglyceride polymerization is the method currently used by NatureWorks to produce high molecular weight PLA polymers. Propyl esters are cyclic dimers of milk, which are intermediate products in the ring-opening polymerization of PLA. First prepolymerization of D-lactic acid, L-lactic acid or a mixture of both to obtain intermediate lactate oligomer chains (<1000 lactate repeating units), followed by catalytic reactions at lower pressures to depolymerize and obtain a mixture of propanediol ester stereoisomers. Propanediol, the cyclic dimer of lactic acid, is formed by the condensation reaction of two lactic acid molecules and has three stereospecific forms, as shown in Figure 4-3: L-propanediol (two L-lactic acid molecules), D-propanediol (two D-lactic acid molecules) and racemic propanediol (one L-lactic acid and one D-lactic acid molecule). According to Hartmann (1998), the formation of different ratios of propyl cross ester isomers is influenced by the lactic acid isomer feedstock, temperature and catalyst. The propylene glycol ester is optically purified by vacuum distillation and then subjected to native melt polymerization to obtain a high optical purity PLA. Commercial fabricators prefer native melt polymerization because it requires only small amounts of non-toxic catalysts such as low activity metals, carboxylates, oxides and alcohol salts. These efforts contribute to the synthesis of high molecular weight PLAs. It has been observed that in the presence of transition metals (tin, zinc, aluminum, etc.), propyl cross ester readily polymerizes with tin(II) and zinc, which have the ability to produce the purest polymers. Several studies reported that these catalysts would be more effective in propyleneglyceride polymerization due to the presence of covalent metal-oxygen bonds and free p or d orbitals (Kricheldorf et al.; Dahlman et al.).
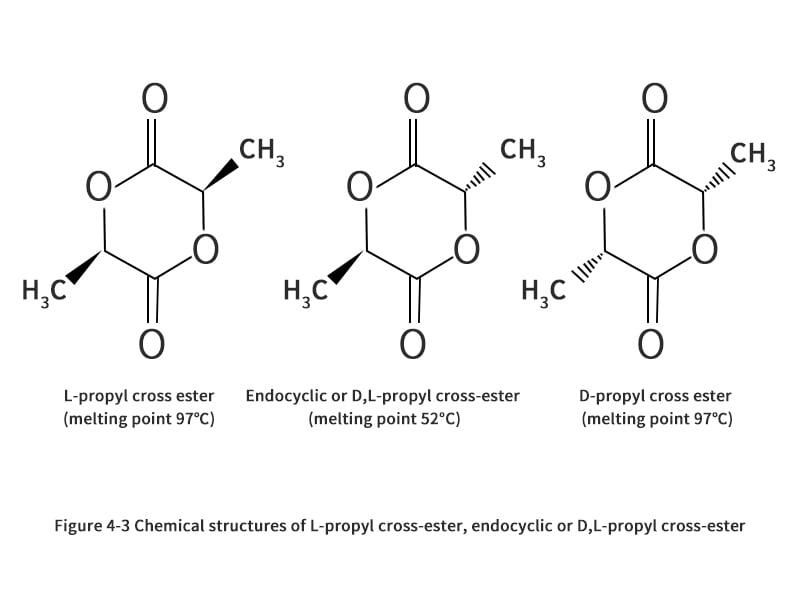
As mentioned earlier, lactic acid is a chiral molecule with L and D dimers, and the composition of these two isomers of lactic acid has a great influence on the properties of PLA. This means that the stereochemistry of PLA can be tailored to its application. The steric regularity of the synthesized monomers determines that PLA is a highly crystalline polymer (H uang et al.). Stereochemically pure PLA can be a crystalline polymer whether it is D-lactic acid or L-lactic acid. Relatively high D or L content (>20%) produces amorphous materials, while highly highly crystalline materials can be obtained when the D or L content is low (<2%) (Lunt, Shafer,). Thus, PLA can consist of three stereoisomers of propyleneglycerides: L-propyleneglyceride, D-propyleneglyceride and racemic propyleneglyceride; depending on the composition, the resulting polymers can have different properties. The stereochemical composition of the polymer has a significant effect on the melting point, the crystallization rate and the final degree of crystallization of the polymer. Drumlight et al. reported that PLA made from pure L-propyl cross-ester has an equilibrium melting point of 207°C and a glass transition temperature of about 60°C. Usually, high stereo chemically pure PLA, whether it is L or D type, has a melting point of about 180°C and an enthalpy of melting of 40 to 50 J/g. Subsequent introduction of stereochemical irregularities into the polymer, such as copolymerization of poly(L-propyleneglyceride) with racemic propyleneglyceride or D-propyleneglyceride, resulted in a significant decrease in melting point, crystallization rate, and degree of crystallization (Figure 4-4), but had no effect on the glass transition temperature (Lunt, 1998). From a study conducted by Kolstad (1996), it was recognized that the peak melt temperature decreases in a roughly proportional manner. For the copolymers with high content of endo-propyleneglycerides, the half-crystallization time of the copolymers was significantly longer (Table 4-1). These findings were further confirmed by Huang et al. , who found that the spherical crystal growth rate was also strongly dependent on the content of the endocyclic riser. The crystallinity of poly(L-lactide-co-meso-lactide) copolymer decreases significantly with the increase of D-isomer content (D-isomer contributed by meso-lactide) , Poly(L-lactide) is 40% to 60%, and the copolymer whose content of meso-lactide (or 6.6% D-isomer) is less than 12% has a crystallinity of less than 20%.
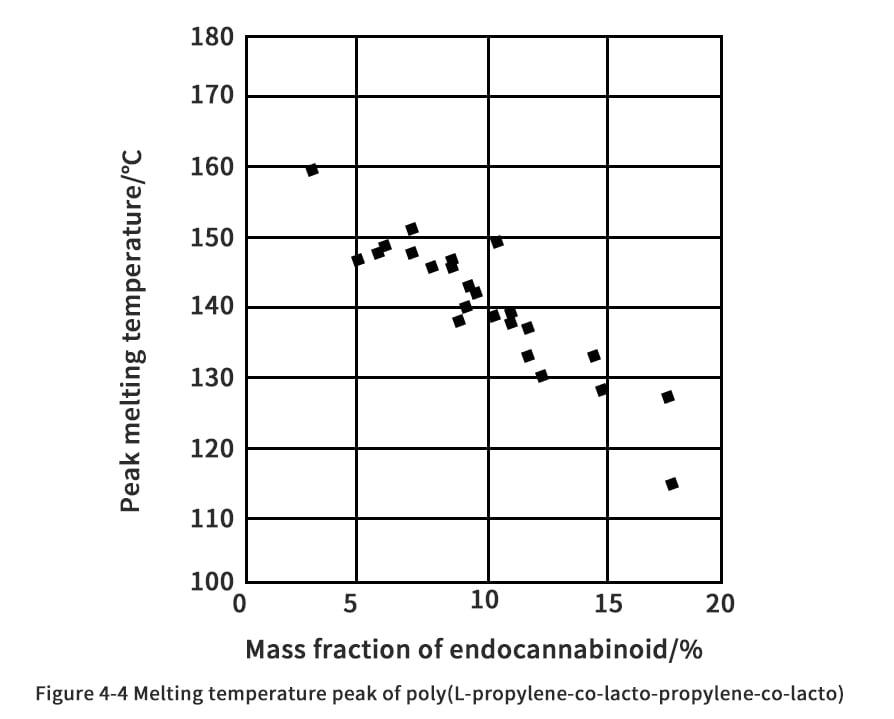
Table 4-1 Semi-crystallization time of poly(L-propylene-co-lacto-propylene-co-lacto) Unit: min
Temperature/°C | 0%endocannabinoid | 0%endocannabinoid | 3% endocannabinoid | 3% endocannabinoid | 6% endocannabinoid | 6% endocannabinoid |
| Mn=101000 | Mn=157000 | Mn=88000 | Mn=114000 | Mn=58000 | Mn=114000 |
85 | 14.8 | — | 23.9 | — | — | — |
90 | 7.0 | 11.4 | 11.0 | — | — | — |
95 | 4.5 | — | 8.1 | — | — | — |
100 | 3.8 | 4.8 | 9.4 | 11.4 | 27.8 | — |
105 | 2.9 | — | 8.6 | — | 19.6 | — |
110 | 1.9 | 4.0 | 6.0 | 10.8 | 19.7 | 44 |
115 | 3.5 | — | 6.9 | — | 22.2 | — |
120 | 4.0 | 5.7 | 8.2 | 11.6 | — | — |
125 | 5.1 | — | 11.5 | — | — | — |
130 | 8.7 | 13.4 | — | — | — | — |
135 | 22.9 | — | — | — | — | — |
Pure crystals of PLA, i.e., 100% crystallinity, have a theoretical enthalpy of melting (ΔHm) of 93.7 J/g, compared to the experimental value of 40-50 J/g for polymers with 37% to 47% crystallinity (Tsuji, Ikada). Note that the degree of crystallization can vary depending on the cooling rate, polymerization conditions, and the presence of impurities or enantiomers. Huang et al. and Nijenhuis et al. reported that the heat of melt can reach 100 J/g during slow polymerization, resulting in highly crystalline stereosymmetric polymers. Depending on the preparation method and thermal history, PLA can crystallize into alpha, beta or gamma types. De Santis and Kavacs found that the conformation of the chain in the α-phase is the left-handed 107-helix L-isomer (PLLA), while the D-isomer (PDLA) is the right-handed 103-helix. Both PLA chains have an orthogonal unit size cell with a="10.7" Å, b="6.126" Å, and c="28.939" Å. Based on the ratio of the a and b parameters of 1.737 (approximately √3), it is arranged in a spiral hexagonal shape. Hoogsten et al. (1990) showed that the β-type of PLA is also present in the orthogonal unit cell with parameters a = 10.31 Å, b = 18.21 Å and c = 9.0 Å, which houses six helices with a subhexagonal arrangement (b/a ratio of 1.76, i.e. √3). In addition, Brizzola et al. studied orthorhombic cells based on the triple helical conformation of two parallel chains and found the existence of two distinct interconnected phases. The γ-crystalline PLLA can be recovered by epitaxial crystallization of two antiparallel s(3/2) helices in a pseudo-rhombohedral crystal system (a=9.95, b="6.25," c="8.8)," showing a triple helical configuration.
4.3 PLA analysis techniques
4.3.1 Nuclear magnetic resonance spectroscopy
PLA is produced by ring-opening polymerization of a cyclic dimer of lactic acid, propylene glycol. In addition to the presence of lactic acid stereoisomers, the properties of PLA are influenced by the number and distribution of the L and D stereocenters of the polymer chain. Nuclear magnetic resonance (NMR) spectroscopy plays an important role in determining the stereo-order distribution of polymers. PLAs with a high degree of stereospecificity are known to form highly crystalline polymers, i.e. isotactic PLAs consisting of D-propanediol or L-propanediol, which have a higher crystallization rate compared to racemic-propanediol, which tend to form amorphous PLAs when synthesized using non-stereoselective catalysts. NMR applies the principle of absorption and re-emission of electromagnetic radiation by nuclei in a magnetic field to explain the flexibility of the structural orientation of polymers.
The NMR spectra show resonances of specific polymers with steric order sensitivity. In the case of PLA, the NMR spectrum can resolve the dimers -LD- (or -DL-) and -LL- (or -DD-). However, for similar dimers, -DD- and -LL-, or -LD- and -DL, did not show different chemical shifts. In the steric order of PLA, -DD- and -LL- produce isosteric pairwise relationships, while -LD- and -DL- have interstitial pairwise structures. There are difficulties in the observation of NMR results, such as superposition of chemical shifts, lack of resolution and the possibility of steric order formation due to the polymer chains staying in a large macromolecule. For the steric order sensitivity of length n, 21 (n-1) possible combinations of pairwise relationships can be observed in the NMR spectrum.
The stereosequence distribution of PLA was studied using NMR spectra.Kricheldorf and Kreiser-Saunders pioneered the use of hypermethyl resonance in the 1H and 13C NMR spectra of various PLA synthesis methods and the initiators/catalysts involved. Zell et al. revised the four-dimensional stereo-order assignment of methane carbon and protons of Kricheldorf et al. The modified four-dimensional stereo order is an extension of the methyl stereo sequence assignment used in PLA for upgrading to the six-dimensional level and includes methods for quantifying the L-propyl cross-ester, D-propyl cross-ester, and racemic propyl cross-ester content in PLA.
4.3.2 Infrared Spectroscopy
Infrared (IR) spectroscopy is an analytical method used to determine the presence of functional groups and to reveal the bonding or interactions within a substance. The IR spectra of polymers are usually analyzed using the Fourier transform infrared (FTIR) method, with scans typically performed at 4000 to 400 cm-1 and results expressed as transmittance or absorbance. IR spectra can capture bond vibrations and provide evidence to identify functional groups. Stronger bonds are usually more rigid and require more force to stretch or compress them.
Recently, Pan et al. investigated the crystal structures of PLLA and PLLA/PDLA stereocomplexes using FTIR technique. PLA will form different crystal forms under different crystallization conditions. It is usually α-type and crystallizes by cold crystallization, melt crystallization or solution route, producing unitary crystals in a distorted 103 conformation of the rhombohedral crystal system (pseudo-rhombohedral system) (Aleman et al.). When α-crystals are stretched to high stretching ratios at high temperatures, PLA transforms to β-type, which adopts a 31-helical conformation (Sawai et al.). At lower crystallization temperatures Tc (<100 °C), sterically regular PLA melts crystallize to give another sub-stable α' type, while at higher Tc (>120 °C) α types are obtained (Zhang et al., 2005a). α PLAs were found to be spectrally split by Pan et al. (2011).
When cooled to 140°C, the α-type PLA split into several new peaks: 3006 cm-1 (CH3 asymmetric stretching), 2964 cm-1 (CH3 symmetric stretching), 1777 cm-1 and 1749 cm-1 (CO stretching), 1468 cm-1 and 1443 cm-1 (CH3 asymmetric bending), 1396 cm-1 and 1381 cm-1 (CH3 symmetric bending), 1222 cm-1 (COC asymmetric bending and CH3 asymmetric oscillation), 1144 cm-1 (CH3 asymmetric oscillation) and 1053 cm-1 (CH3 asymmetric oscillation). 1 (CH3 symmetric bending), 1222 cm-1 (COC asymmetric bending and CH3 asymmetric oscillation), 1144 cm-1 (CH3 asymmetric oscillation) and 1053 cm-1 (CCH3 bending). The α' crystals show significant results compared to the former without spectral splitting. This is because α' crystals have weaker interchain interactions in their lattice. In other words, there is a lack of lateral interactions between the molecular chains in the crystal unit cell. Comparison with the amorphous structure of PLLA/ PDLA blends revealed that the corresponding CO stretching peaks of the blends were about 10 cm-1 lower, while CH3 asymmetric/symmetric stretching and CH symmetric stretching were reduced by 4-6 cm-1. Therefore, it can be assumed that CHOC forms weak hydrogen bonds in amorphous PLA crystals (Zhang et al.).
Paragkumar et al. (2006) performed FTIR analysis of poly(D,L-pharma-cross-ester) and poly(D,L-propyl-cross-ester-co-ethyl-cross-ester) films to characterize the surface bias in films of different thicknesses. They found that the attenuated total reflection (ATR)-FTIR and FTIR transmission spectra of poly(D,L-propyleneglycolate) and poly(D,L-propyleneglycolate-co-ethyleneglycolate) films were very similar to the variation of some peak intensities. They compared and discussed the peaks with significant changes in intensity.
4.4 Solubility and barrier properties of polylactic acid
PLA is a bio-based polymer suitable for replacing traditional petrochemical polymers as packaging materials. The "green" nature of PLA has been favored by the food packaging industry because of its excellent barrier properties that keep food fresh while not polluting the environment. Food manufacturers must choose their packaging materials carefully in order to avoid chemical and biological contamination and to avoid accelerated deterioration of food. Packaging materials must provide an adequate barrier to water vapor to prevent food degradation or microbial growth, prevent oxidation due to gas penetration in the atmosphere that may be triggered, and maintain the volatile organic compounds contained in the food to preserve aroma and flavor. In addition, the package should be insoluble in a variety of solvents to avoid the transfer of trace substances from the package to the food, which may be hazardous to health after consumption.
In general, the likelihood of food contamination or poisoning from commercially available PLA containers is very low. This is because PLA is produced from propanediol monomer, which is derived from L-lactic acid, a non-toxic component that occurs naturally in the human body. However, trace amounts of D-lactic acid may be present during the polymerization process, which is a minor by-product. Due to the lack of appropriate enzymes, D-lactic acid cannot be consumed by the body. The determination of the permeability (solubility and diffusivity) of gases, flavors and aromas in polymers is essential for the application of PLA in the food packaging industry. This will be discussed in the next section. In Chapter 2, Synthesis and Production of PLA, methods for the determination of D-lactate and propanediol content for safety purposes are discussed, which is an important aspect of the application of PLA as a packaging material.
4.4.1 Solubility of poly(lactic acid)
According to Nampoothiri et al., PLA is soluble in chloroform, dichloromethane, dioxane, acetonitrile, 1,1,2-trichloroethane, and dichloroacetic acid. PLA is also soluble in toluene, acetone, ethylbenzene and tetrahydrofuran (THF) when heated to boiling temperature, but has limited solubility at low temperatures. In general, PLA is insoluble in water, some alcohols and all alkanes. Highly crystalline PLLA is resistant to solvents such as acetone, ethyl acetate and THF, while amorphous PLA, such as copolymers of poly(D,L-propyleneglycerides), can be readily dissolved in various organic solvents such as THF, chlorinated solvents, benzene, acetonitrile and dioxane.
The solubility of PLA depends on the crystallinity of the polymer, as the highly oriented structure increases the difficulty of inter-molecular chain migration of the solvent. The thermodynamic criterion of solubility is based on the free energy of mixing (ΔGm), which indicates that two substances are mutually soluble if ΔGm is zero or negative. The mixing free energy of the dissolution process between solvent and polymer is related to △Gm=△Hm-T△Sm, where △Hm, T and △Sm are the mixing enthalpy, absolute temperature and mixing entropy, respectively. Usually, the value of △Sm is small and positive. Thus, the solubility of a solvent depends strongly on ∆Hm and T。. The solubility of a substance is expressed by the solubility parameter (δ), which was introduced by Hildebrand and Scott and is related to the cohesion energy density. Hansen and Skaarup later proposed solubility parameters related to polar and hydrogen bonding systems, which were classified as nonpolar (δd), polar (δp), and hydrogen bonding (δH), where Hansen's solubility parameter δt = δd + δ + δk。 Table 4-2 summarizes the solubility parameters of the solvents. In order to dissolve PLA in the solvent, the solubility parameters of the polymer and the solvent should differ by δt < 2.5 (Auras). The solubility of liquid components contained in food, such as water, ethanol and paraffin (represented by hexane), differs more significantly from the solubility parameters of PLA; therefore PLA is safe in contact with food without transfer.
Table 4-2 Solubility parameters of solvents at 25°C (HANSEN, 2000)
4.4.2 Permeability of poly(lactic acid)
When used as a packaging material, the gas permeability properties of PLA are very important. Packaging requires low permeability materials to avoid loss of flavor and aroma or oxidation, thus extending the shelf life of the food. Since PLA is a biodegradable material that has the potential to replace existing plastic materials such as PET, PS and low density polyethylene (LDPE), it is important to have permeation properties that are as effective as those existing polymers.
Lehermeier et al. investigated the gas permeability of PLA to nitrogen, oxygen, carbon dioxide and methane. The results are summarized in Table 4-3. The permeation activation energy (Ep can be calculated by the following equation:
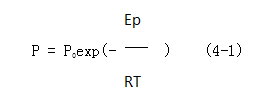
Table 4-3 Parity properties of polylactic acid (PLA) and polyethylene terephthalate (PET)
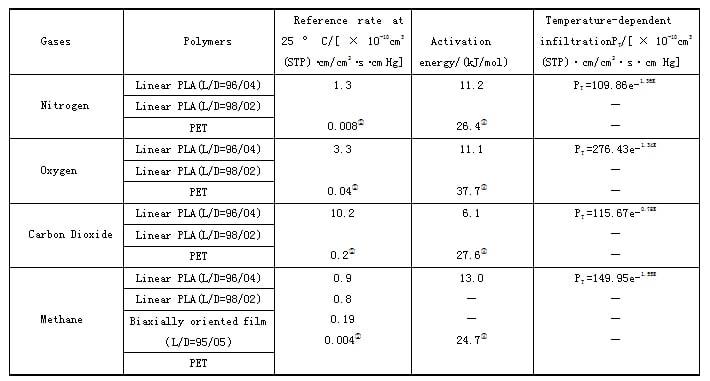
①Michaels(1963)。
②Pauly(1990)。
It can be seen that the permeability of PET is lower than that of PLA, in other words, PET has better barrier properties than PLA, with an L:D of 96:4. Lehermeier et al. concluded that this is due to the reduced free volume and chain mobility of PET containing aromatic rings in the polymer chain backbone. The introduction of branched chains in the PLA chain does not result in significant changes. However, crystallization can greatly improve the barrier properties. In the biaxially oriented PLA film with 16% crystallinity (L:D of 95:5), the increase in crystallinity reduced the permeability of this film to two-ninths of the permeability of PLA films with 1.5% and 3% crystallinity (L:D of 96:4 and 98:2), respectively. This is because the crystallinity increases the denseness of the structure, resulting in difficulty in diffusion of gas molecules through the film. PLA has better barrier properties than PS and LDPE. PLA has been shown to be a good barrier to nitrogen, carbon dioxide and methane, but a slightly weaker barrier to oxygen. This finding is important because it suggests that PLA can be used as a powerful packaging material to replace a variety of commercial petroleum-based plastic films. PLA's excellent barrier properties, along with biodegradability and "green" production, mean that it is a strong contender for the packaging material of the future.
Water permeability is another important factor to consider for packaging materials. Shogren (1997) compared the water vapor permeability of various biodegradable polymers, including poly(β-hydroxybutyrate-co-hydroxyvalerate) (PHBV), poly(ε-caprolactone) (PCL) containing 6%, 12% and 18% valerate. The water permeability of these materials was determined by Shogren and is shown in Table 4-4. Compared to many biodegradable polymers other than PHBV, PLA exhibits good water resistance. In addition, annealing of PLA at 130°C caused the formation of crystalline structures, which improved the water resistance. This can be explained by the fact that crystallization reduces the molecular cross-sectional area required for diffusion and increases the diffusion path length by limiting the mobility of the amorphous phase (Shogren, 1997). Similarly, the solubility parameter of the polymer greatly affects the water vapor permeability. When the difference between the solubility parameter values of the polymer and water is small, it means that the polymer is hydrophilic and, therefore, the rate of water vapor transport is higher for the polymer.
Table 4-4 Water vapor transmission rates of biodegradable polymer films (Shogren, 1997)
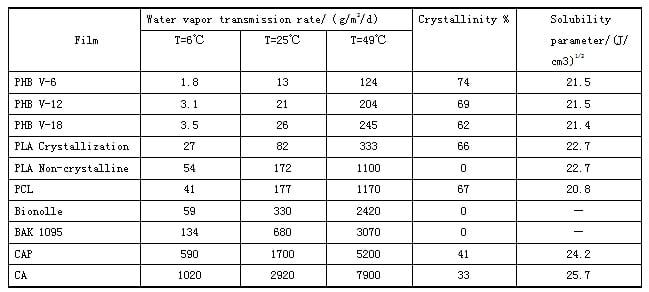
Note: PHBV-poly (β-hydroxybutyrate-co-hydroxyvalerate) with 6, 12 and 18% valerate; PLA crystals-PLA annealed at 130°C;PCL-Poly(ε-caprolactone);CA-Cellulose acetate;CAP-Cellulose acetate;PLA-Polylactic Acid;Bionelle - blown films containing aliphatic polyesters;BAK1095 - Blown film containing poly(ester amide)。
The solubility parameter of water is 47.9(J/cm3)1/2。
Siparsky et al. investigated the effect of copolymerization on the water permeability of PLA membranes in depth. The "solution diffusion" model [Eq. (4-2)] is used to characterize the water vapor in PLA. p is the permeability coefficient related to the flow rate, S is the solubility coefficient representing the equilibrium water concentration, and D is the diffusion coefficient related to the diffusivity.
Solving the diffusion model: P = S × D (4-2)
Table 4-5 shows the water vapor diffusivity as affected by the L/D composition in PLA. The isosteric polymeric isomers of PLA are due to their oriented structure which has better water vapor barrier properties. However, the crystallinity of PLA has no effect on the permeability of water vapor. The incorporation of caprolactone monomer has little effect on the diffusion coefficient shadow, but the incorporation of polyethylene glycol (PEG) causes a dramatic decrease in water vapor resistance. The hydrophilic nature of polyethylene glycols and the disruption of their structure are factors that lead to reduced barrier properties when combined with PLA.
Table 4-5 Diffusion, solubility and permeability coefficients of PLA, PCL and their copolymers/blends measured at 90°C with a relative humidity of 20°C
Composition | Tg/℃ | Crystallinity/% | P/×1013 | S/×106 | D/×106 |
PLA(L/D=50/50) | 52 | — | 2200 | 3400 | 0.067 |
PLA(L/D=70/30) | 50 | — | 2200 | 2200 | 0.10 |
PLA(L/D=90/10) | 54 | — | 1500 | 2000 | 0.078 |
PLA(L/D=95/5) | 59 | — | 1400 | 3000 | 0.044 |
PLA(Hardened)(L/D=100/0) | 63 | 11 | 1900 | 4000 | 0.052 |
PLA(L/D=100/0) | 63 | 39 | 1600 | 4000 | 0.046 |
PLA(Annealed at 160°C for 15min, | 63 | 46 | 2000 | 4000 | 0.040 |
(L/D=100/0) | 63 | 46 | 2000 | 4000 | 0.040 |
30% random copolymer PCL-PLA | 40 | PCL<5 | 2900 | 2200 | 0.13 |
30% block copolymerPCL-PLA | —63,47 | PCL;9 | 3100 | 3100 | 0.10 |
30% orientation PCL-PLA | —63,73 | PCL;11 | 2700 | 2600 | 0.11 |
PCL | —60 | 52 | 3200 | 1600 | 0.20 |
20% co-mingling PEG-PLA | 48 | — | 5700 | 10900 | 0.052 |
Note: P unit is cm3 (STP) cm/cm2s Pa. S unit is cm3 (STP) cm3 Pa. D unit is cm2/s.
4.5 Conclusion
The properties of PLA are strongly influenced by the stereochemistry of its monomer. When PLA has high stereochemical purity, it tends to form a highly crystalline crystal structure. Copolymerization with different propyleneglycolate isomers can yield a variety of PLA properties. The effect of isomerization on PLA can be detected by IR and NMR spectroscopy. Many studies have demonstrated the low solubility of PLA in various solvents/liquids such as water, alcohols and paraffins. This indicates that PLA can be safely used as a food packaging material without causing adverse health effects. In addition, PLA has the same effective barrier properties as LDPE and PS. The "green" qualities of PLA mean that it is a viable environmentally friendly alternative to petroleum-based polymers.
5. Mechanical properties of polylactic acid
5.1 Introduction
Depending on parameters such as crystallinity, polymer structure, molecular weight, material formulation (blends, plasticizers, composites, etc.) and orientation, the mechanical properties of commercial polylactic acid (PLA) can vary from soft, elastic materials to hard, high-strength materials.
PLA, also known as polypropylene cross-ester (polylactide, the polymerization of cyclic lactic acid, which is also known as propylene cross-ester), is a brittle material with low impact strength and elongation at break, more similar to another relatively brittle polymer, polystyrene (PS). The poor toughness of PLA limits its use in applications where plastic deformation is required at high stress levels. In order to improve the mechanical properties of PLA, especially the toughness, various modification methods have been used.
5.2 Effect of crystallinity and molecular weight of PLA on mechanical properties
In terms of structural property relationships, crystallinity is an important property that affects the mechanical properties of PLA (Figure 5-1). Perego et al. investigated the effects of molecular weight and crystallinity on the mechanical properties of poly(L-propyleneglycolate) (PLLA), poly(D,L-propyleneglycolate) (PDLLA) and annealed poly(L-propyleneglycolate) (Ann. PLLA). They reported that PL LA and PDLLA with different molecular weights showed slight variations in tensile strength, ranging from 55 to 59 MPa for PLLA and 40 to 44 N MPa for PDLLA. the results are shown in Tables 5-1 and 5-2.
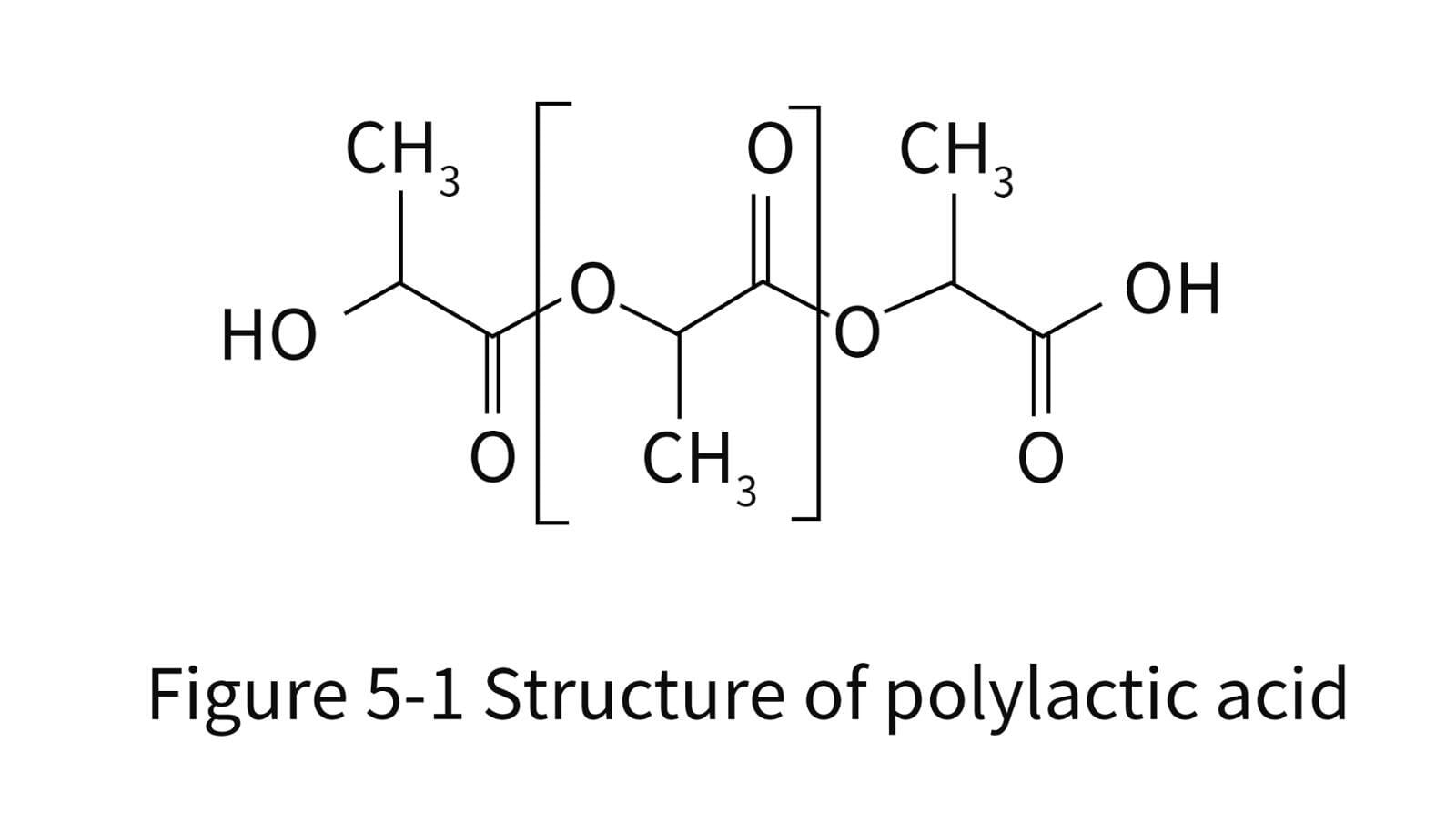
Table 5-1 Mechanical properties of poly(L-propyleneglycolate) (PLLA) samples
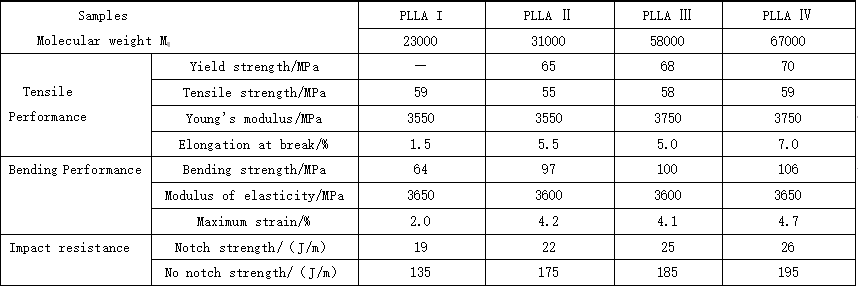
Table 5-2 Mechanical properties of non-annealed poly(D,L-propyleneglycolate) (PDLLA) specimens
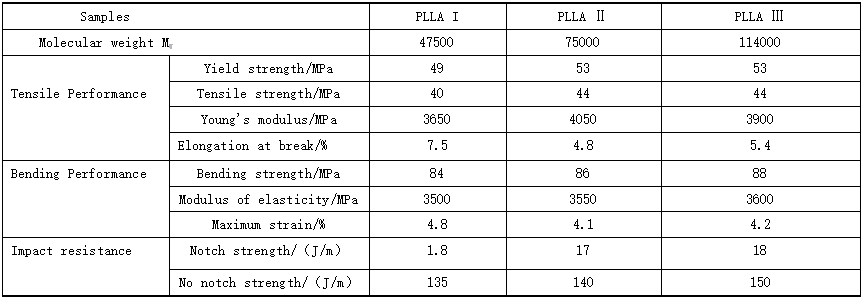
However, as shown in Tables 5-1 and 5-2, PLLA shows better strength than PDLLA with Mw = 67000 with a strength of 59 MPa; while PDLLA III with Mw = 114000 has a strength of 44 MPa. This enhancement is thought to be due to the steric regularity of the polymer chains. In other words, the presence of L and D stereoisomers in PLA affects the crystallinity and structural chain alignment, which leads to changes in physical and mechanical properties.
Table 5-3 shows the effect of annealing on the mechanical properties of PLLA. The tensile strength increases slightly from 47 MPa to 66 MPa over the molecular weight range of annealed PLLA. Obviously, the crystalline part of this material corresponding to the crystallinity is influenced by the molecular weight increase. The highest tensile modulus of elasticity was obtained for the annealed PLLA samples in the range of 4000-4200 MPa, while the tensile modulus of elasticity for the non-annealed PLLA samples ranged from 3550-3750 MPa. Similar results can be observed for the bending strength, where the annealed PLLA samples have higher bending strength than the unannealed PLLA and PDLA samples. The trends in tensile and flexural strengths show that these properties increase with increasing crystallinity, especially when the Mn of annealed PLLA samples is above 55000. The impact resistance of PLLA varies with the molecular weight and is higher after annealing due to the rigidity effect of the crystal region. However, the impact strength of PDLLA samples is not related to the molecular weight size because of its completely amorphous structure.
Table 5-3 Mechanical properties of annealed poly(L-propyleneglyceride) (Ann.PLLA) samples
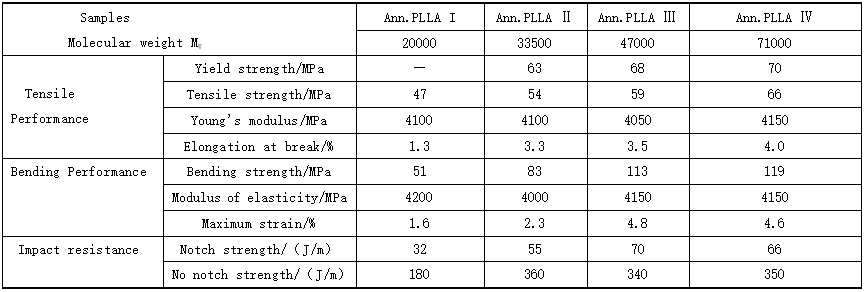
5.3 Effect of modifiers/plasticizers on PLA
Lactic acid is a glassy polymer with low elongation at break (<10%). Typical biodegradable and non-biodegradable plasticizers have been used to lower the glass transition temperature, increase ductility, and improve processing properties (Mascia, Xanthos, 1992). This has been achieved by controlling the molecular weight, polarity and terminal groups of the plasticizer added to the PLA.
Propylene glycol is an effective monomer for plasticizing PLA. The elongation at break was increased to 288% by adding 17.3% propyleneglyceride to PLA. However, it has the disadvantage of rapid migration and loss, resulting in hardened polymers and sticky surfaces (Sinclair, 1996). In contrast, high molecular weight plasticizers are not likely to migrate, so they are used by the researchers. Tables 5-4 summarize the mechanical properties of PLA after using different plasticizers.
Table 5-4 Summary of reported mechanical properties of plasticized PLA
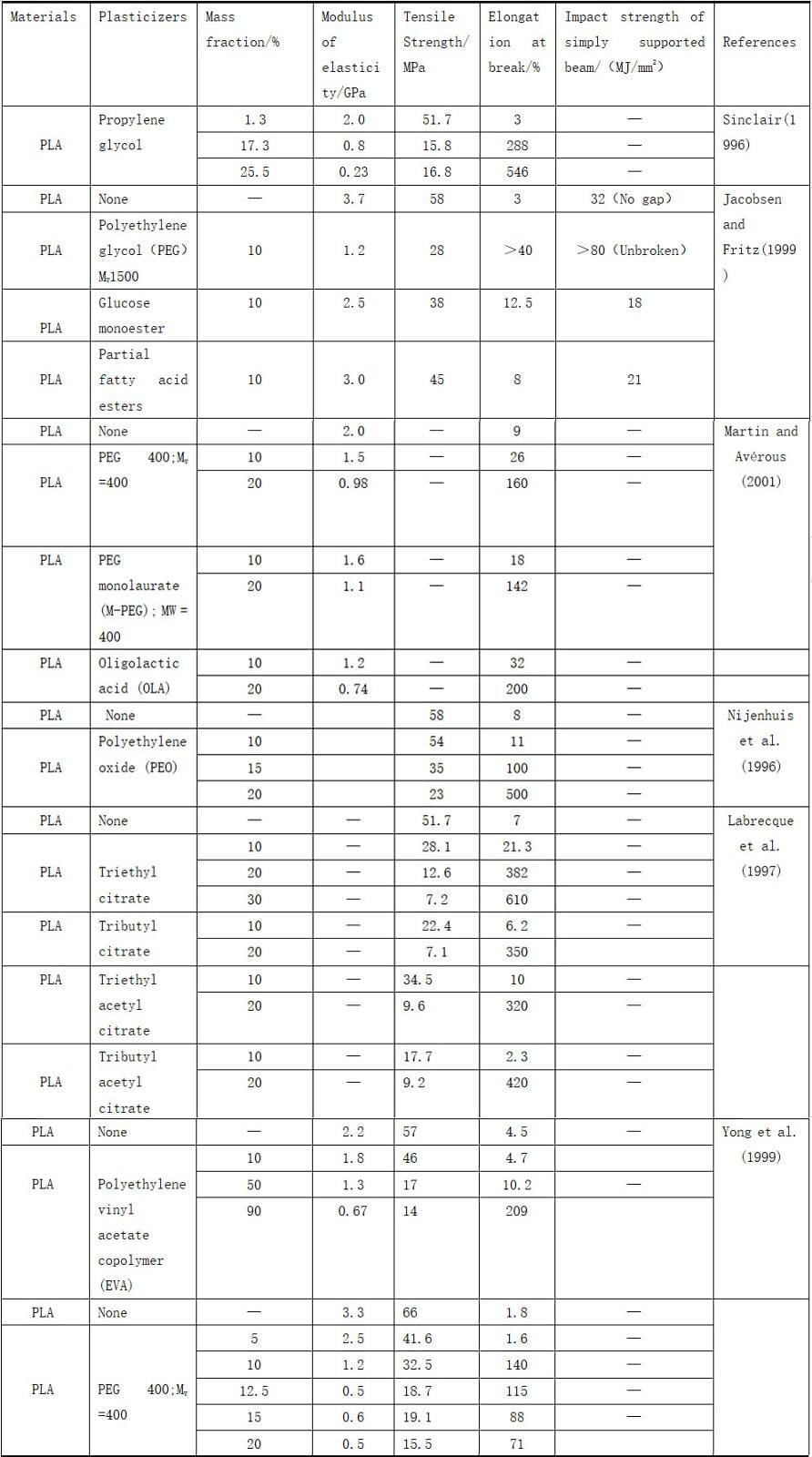
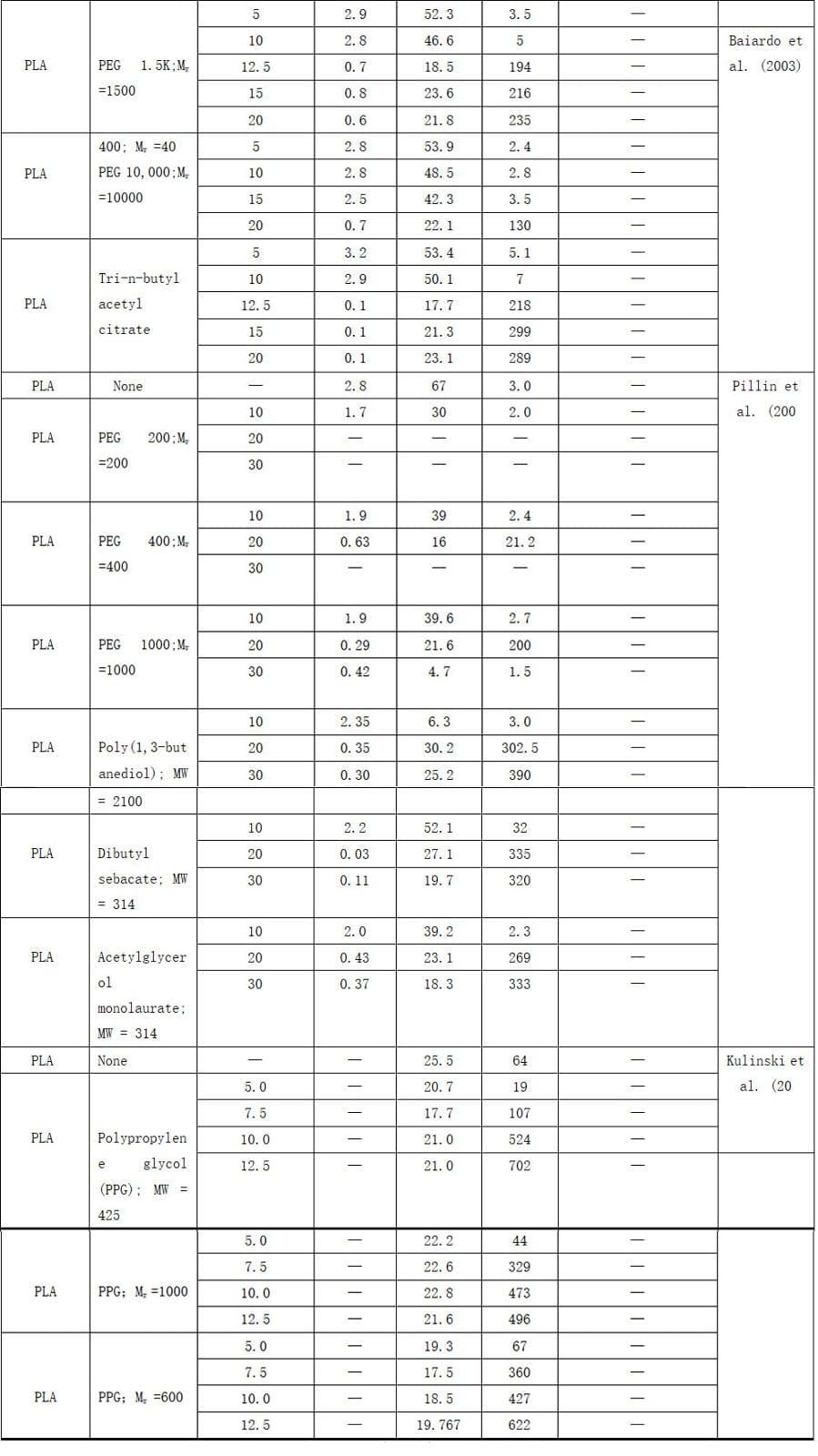
Jacobsen and Fritz studied the effect of three different types of plasticizers on PLA, namely polyethylene glycol (PEG 1500; Mw = 1500), glucose monoester and partial fatty acid esters to compare their properties. They observed that, in general, the addition of different types of plasticizers resulted in a decrease in the elastic modulus. The addition of 2.5% (all refer to mass fraction in this chapter) can reduce the modulus by 10% to 15%. The reduction in modulus is more pronounced when the plasticizer is added in larger amounts (5% and 10%). Similar results were obtained for tensile strength: tensile strength decreased gradually with increasing PEG and glucose monoester content, and linearly or slightly with increasing partial fatty acid ester addition.
For elongation at break, an increase in the partial ester content of fatty acids leads to a decrease in this value. This is because some of the fatty acid esters after sufficient dispersion can excite crack formation. However, the remaining two plasticizers showed the opposite effect on some fatty acid esters, i.e., the elongation at break increased with increasing plasticizer dosage. In terms of elongation. PEG is the best plasticizer, and when added at 10%, the elongation at break of PLA can be increased by 180%.
Neither the addition of glucose monoesters nor some fatty acid esters improved the impact resistance of PLA at any concentration. In fact, they reduce the impact strength due to the interference generated by the plasticizer particles in the PLA matrix that limits the chain sliding that absorbs the impact energy. This effect was also observed at low levels of PEG in PLA. Small amounts of PEG lead to a decrease in impact resistance, but at a concentration of 10%, the plasticizing effect dominates The impact resistance increases substantially to the point where no fracture is observed.
Several authors have reported the use of polymeric plasticizers to improve performance. Nijenhuis et al. found an improvement in elongation at break by incorporating high molecular weight polyethylene oxide (PEO) into PLLA. The effect is most pronounced when the PEO concentration exceeds 10%. For example, at a PEO concentration of 20%, elongation at break was up to 500%, however, as expected, the tensile strength at this point was reduced from 58 MPa for pure PLLA to 24 MPa. Labrecque et al. investigated the performance of citrate esters generated from natural citric acid as PLA plasticizers. All the plasticizers added significantly reduced the tensile strength of PLA (about 50%), and even at 10%, the reduction in tensile strength of PLA was greater with increasing plasticizer concentration. However, while elongation at break increased significantly at higher concentrations (>20%) in all cases, there was no significant change in elongation at break at lower concentrations (<10%). The maximum value of elongation at break was 610% when triethyl citrate was 30%, but at the same time the tensile strength was significantly lower.
Yoon et al. investigated the role of ethylene vinyl acetate (EVA) as a plasticizer in PLLA. They found a slight increase in the elongation at break of the PLLA/EVA blends up to 70% of the EVA. However, the elongation at break of the blends increased significantly (209%) when the EVA content was 90%. However, with the increase of EVA content, the tensile strength and modulus of PLLA -EVA blends first decreased rapidly, and then decreased slowly. Martin and Avérous used PEG, PEG monolaurate and low polymeric lactic acid to plasticize PLA. They found that the addition of these plasticizers reduced the modulus, which ranged from 28% to 65% depending on the type and concentration of plasticizer used. The addition of 20% polyethylene glycol (Mw = 400) and low polymeric lactic acid reduced the modulus of elasticity by 53% and 65%, respectively. At the same time, the elongation at break increases with the increase of plasticizer concentration. Elongation at break is as high as 200%, indicating that the properties of PLA can easily change from rigidity to toughness. Baiardo et al. (2003) used tri-n-butyl acetyl citrate and PEG of different molecular weights (Mw between 400 and 10,000) to plasticize PLA. These researchers also observed a significant increase in elongation at break at the expense of strength and tensile modulus. Examination of the elongation at break values showed a twofold change in elongation at 5% plasticizer content, but also depending on the type of plasticizer used. At Mw = 10000 for PEG, 20% PEG is required to induce a large increase in elongation at break, but using only 10% low molecular weight PEG (Mw = 400) causes the same change.
Ren et al. (2006) reported that the polyplasticizers low molecular weight glycerol triacetate and oligomeric poly(1,3-butanediol adipate) were also used to plasticize PLA. They found that this achieved significant improvement in elastic properties, but at the cost of tensile strength. Elongation at break tended to level off when the plasticizer content was 0 to 5%, while it increased sharply when the plasticizer content was 5% to 9%. The results show that when the plasticizer content is less than 5%, the blends are brittle, and when the plasticizer content is greater than 9%, the blends are tough.
Pillin et al. investigated the effects of polyethylene glycol (PEG) (Mw range 200-1000), poly(1,3-butanediol) (PBOH), dibutyl sebacate (DBS) and acetylglycerol monolaurate (AGM) of different molecular weights as plasticizers on PLA. Young's modulus decreases sharply when the plasticizer content is greater than 20%. Compared with other plasticizers, PEG makes the Young's modulus drop even lower. However, the physical and mechanical properties of PLA were not deteriorated when the Mw = 200 content of PEG was 10%, or when the Mw = 400 content was 20%, or when the Mw = 1000 content was 30%. When the plasticizer content was high, the material became friable due to the lack of adhesion between the separated phases. Therefore, the plasticizer efficiency of plasticizers is related to the compatibility at the molecular level, from which PEG is more compatible than other molecules. The elongation at break increased as the amount of plasticizer increased, but the optimum addition of PEG was 20%, while the optimum addition of other plasticizers was greater than 20%. That is, PLA blends of plasticizers PBOH, AGM and DBS have higher cohesive power than PEG. At 20%, the most effective plasticizer was AGM, which reduced the modulus of elasticity value from 2840 MPa to 35 MPa. The mechanical properties of PBOH and DBS were better than those of PEGs, and the resulting materials were not brittle. When large amounts of plasticizers (PBOH, AGM and DBS) are mixed with PLA (about 30%), the tensile modulus or elongation at break is stable relative to the 20% content, but the tensile strength is slightly reduced at this time. Based on these results and mechanical requirements, it is clear that the most effective plasticizing formulations are 20% to 30% AGM, PBOH and DBS.
Kulinski et al. investigated the characteristics of PEG and propylene glycol (PPG) when used as PLA plasticizers. The advantages of using PPGs are their non-crystallization, low glass transition temperature, and miscibility with PLA. The tensile strength and average elongation at break of pure PLA were 26 MPa and 64%, respectively, when PLA was plasticized with PPGs with Mw of 425 and 1000. Starting from 7.5% plasticizer content, the elongation at break of all blends exceeded that of pure PLA, and the peak elongation at break reached 500% to 700% when the plasticizer content reached 12.5%. The higher the content of PPG and the smaller the molecular weight of PPG, the more pronounced this effect becomes. However, the deformation indicates that the strength of the blends is usually lower than that of pure PLA, in the range of 17.5 to 22.8 MPa. As shown in Table 5-4, the 12.5% low molecular weight PPG was the most effective as a PLA plasticizer because it resulted in the greatest increase in elongation at break and the least decrease in tensile strength.
5.4 Polylactic acid blends
Polymer compounding is another way to obtain newmaterials with desirable properties, based on commercial polymers rather than designing and synthesizing entirely new polymers. Since the 1980s, the development of commercial polymer blends has been rapid and research in this area has remained hot. Mixing different polymers and maintaining their respective properties in the final mixture is a very attractive and inexpensive way to obtain new materials. The preparation of blends usually involves the use of twin-screw extruders, and many factors must be considered in order to obtain a blend with good properties. For example, the barrel temperature must be set above the glass transition temperature of the amorphous polymer component and above the melting point of the semi-crystalline polymer component to obtain optimal dispersion at the appropriate viscosity. For PLA blends, the lower limit should be around 180°C. If a polymer requires a very high processing temperature (greater than 270°C).
then it will lead to thermal degradation of PLA and will not be suitable for blending with PLA.
Achieving the desired results through polymer blending is not always easy. A number of problems arise when dealing with incompatible blends, the most obvious of which is how to obtain good interfacial adhesion between the blended phases, which will directly affect the morphology of the blend and thus the physical and mechanical properties of the blend. If the added polymer is not very compatible with the PLA, significant subsequent development work will be required to improve the compatibility. Poor interfacial bonding can lead to embrittlement of the product, and the morphology of the phases can change dramatically depending on the processing conditions and the design of the part being produced. Some polymers are not biodegradable and their blending with PLA can affect the compostability of the product. In general, PLA blends can be divided into two categories: degradable polymer blends and non-degradable polymer blends. However, in order to maintain the biodegradability of PLA, research efforts have focused on the blending of PLA with degradable or renewable resource polymers.
The blends of polycaprolactone (PCL) and PLA have been extensively studied by many scholars. This is because PCL has rubbery properties with an elongation at break of about 600% (Wang et al), and thus can be a good candidate for toughening PLA. In addition, PCL is a biodegradable polyester, which means that it can be blended with PLA to obtain a fully degradable material.
Unfortunately, many researchers have found that blends of PLA and PCL, while increasing elongation at break, typically decrease the tensile strength and modulus of the blend. For example, HiljanenVainio et al. (1996) reported that modification of PLLA with 20% PCL resulted in a decrease in tensile modulus, tensile strength, and shear strength, with only a slight increase in elongation at break (from 1.6% to 9.6% for pure PLLA). In contrast, the elastomeric polycaprolactone/L-propyleneglycolate (PCL/L-LA) copolymer with PLLA resulted in significantly higher elongation at break (>100%) compared to pure PLLA and the binary blend. They also found that PLA blends with 5%, 10% and 20% PCL/L-LA copolymer content exhibited yield deformation. Moreover, when the PCL/L-LA copolymer content reaches 30%, the blend exhibits a tough rubber-like behavior. The initial impact strength of PLLA was poor, and when 20% PCL/L-LA copolymer was added, the impact strength was increased by a factor of 4.
Tsuji and Ikada investigated the tensile data of PLA/PCL blended films prepared by solution casting method with dichloromethane as solvent. Despite the increase in elongation at break for the 15% PCL blend, the calculated standard deviation was quite high (250% to 200%). Wang et al. showed that the elongation at break of PLA/PCL reaction blends with triphenyl phosphite as the catalyst was significantly higher than that of pure PLA at a certain fraction (PLA/PCL= 80/20 or 20/80). These results indicate that reactive blends are an effective method for improving the elongation and toughness of PLA. The elongation increased to 127% compared to 28% for the non-reactive binary blends. Also, Maglio et al. (1999) found that when PLLA-PCL-PLLA triblock copolymers were used as bulking agents for PLLA/PCL (70/30 mass ratio) blends, elongation at break (53% vs. 2%) and notched impact strength (3.7 kJ/m2 1.1 kJ/m2 ratio) were improved.
Rroz et al. studied the dissolution of PLA and PCL binary blends with a total polymer mass fraction of 10% in dichloromethane. They found that elongation at break increased significantly only at PCL contents greater than 60% and lost its practical value because it was accompanied by a significant loss of modulus and tensile strength. However, Tsuji et al. observed that the addition of PLLA-PCL diblock copolymers to binary PLLA/PCL blends resulted in improved mechanical properties. The addition of the copolymer increased the tensile strength and Young's modulus of the blend at XPLLA of 0.5 to 0.8, and also increased the elongation at break for all XPLLA values [XPLLA = mass of PLLA/(mass of PLLA and PCL)]. These results strongly suggest that PLLA-CL is compatible with PLLA and PCL, and that PLLA-CL increases the compatibility between the two phases in a blend of PLLA-rich and PCL-rich phases.
Semba et al. reported another reactive blend for the preparation of PLA/PCL blends using diisopropyl peroxide (DCP) as a cross-linking agent.DCP was added to the system to improve the elongation at break of the blends. The optimal ratio of PLA/PCL blends is 70/30. The results showed that the elongation at break peaked at low concentrations of DCP [about 0.2 parts (by mass)]. At low DCP content, specimens tested in tension exhibited yield point and plastic behavior. The composite has 2.5 times the impact strength of pure PLA and good toughness, indicating plastic deformation at its fracture surface. This is an interesting radical-based crosslinking application in PLA blends.
Yuan and Ruckenstein synthesized semi-interpenetrating polyurethane/PLA networks. Polyurethanes were prepared from diols, tertiary alcohols and toluene diisocyanate. The results showed that the optimum ratio of cross-linked polyurethane network blended with PLA was 5%. Compared with 1.6 MJ/m3 of pure PLA, the elongation at break increased to 60% and tensile toughness increased to 18 MJ/m3 .
Grijpma et al. studied the blends of PLA with already lactone (CL) and trimethyl carbonate (TMC) rubber copolymers [poly(TMC/CL)]. They report an increase in notched Izod impact strength from 40 J/m to a maximum of 520 J/m with the addition of 20% copolymer to pure PLA. However, for the homopolymer-polymethyl methacrylate (TMC) and PLA blends, the same phase mass fraction of the rubber phase did not improve the notched impact strength. Joziasse et al. studied the homopolymerization of PLA with poly(trimethyl carbonate) [poly(TMC)] rubber copolymers. They found that samples containing 21% poly TMC rubber blocks in PLA did not rupture in the no-notch impact test. A diblock copolymer of L-propyl cross-ester and caprolactone [P(LA/CL] was blended with PLA to determine the effect of its content on the mechanical properties. The unnotched impact strength of the blends was increased from 5 kJ/m2 to 50 kJ/m2 by adding 20% diblock copolymer.
Hasook et al. reported the mechanical properties of PLA/PCL and organoclay nanocomposites. The results showed that the addition of organic clay increased the Young's modulus of PLA but decreased the strength and elongation at break. Initially, the Young's modulus of the material decreased with the addition of PCL. However, the tensile strength and elongation at break of PLA/organic clay nanocomposites increased with the addition of PCL. The tensile strength of PLA/clay nanocomposites was greatest when PCL (Mw = 40,000) was used.
Chen et al. observed that the addition of small amounts of surfactants (i.e., ethylene oxide and propylene oxide copolymers) increased elongation at break, but other mechanical properties such as tensile strength and modulus also decreased at the same time. In addition, the addition of a small amount of PLA-PCL-PLA triblock copolymer (about 4%) to the PLA/PCL (70/30) blend improved the dispersion of PCL in PLA and enhanced the plasticity of the resulting terpolymer. The elongation at break of the PLA/PCL (70/30) blend increased from 2% to 53% for the ternary blend (Maglio et al.). This result has been shown to be brought about by the good dispersion of the PCL structure, where the PCL phase was reduced from 10 to 15 μm to 3 to 4 μm after the addition of the triblock copolymer (4%), according to the statistics of scanning electron microscopy (SEM) photographs of the fractured surface of the mixture liquid nitrogen. Table 5-5 summarize the mechanical properties of the PLA and PCL blends in the above study.
Table 5-5 Summary of mechanical properties reported for blends of polylactic acid (PLA) and polycaprolactone (PCL)
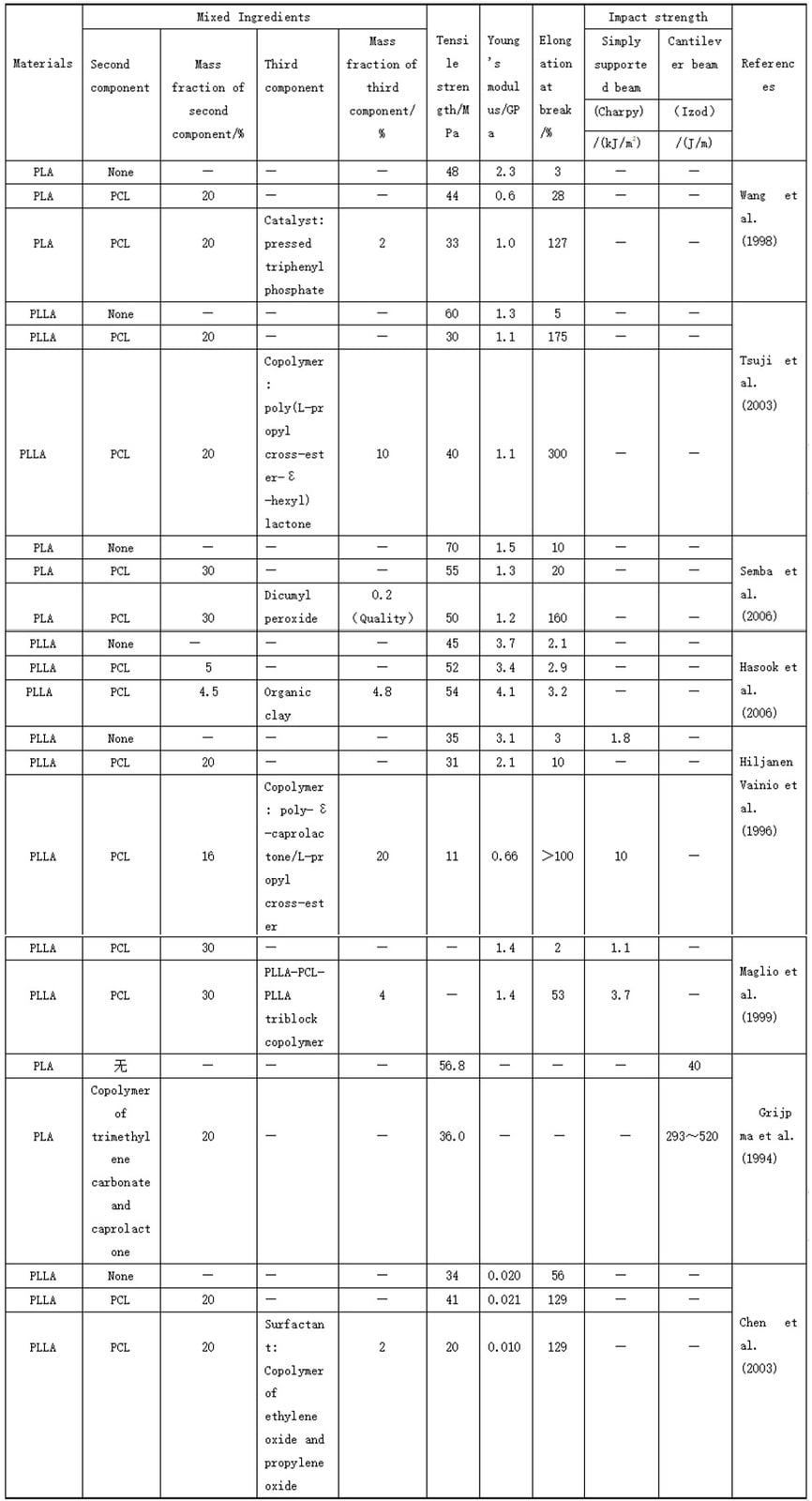
5.5 Blends of PLA with degradable or partially degradable polymers
In addition to PCL, other biodegradable/renewable resource-based polymers have been explored as blends with PLA. For example, Pezzin et al. prepared a mixture of PLA and poly(p-dioxanone) (PPD), which is also a biodegradable polyester. They found that the Young's modulus (1.6 GPa) and elongation at break (55%) of the blends were higher than those of pure PLLA and PPD when only 20% of PPD was added to the PLLA phase, but the tensile strength was lower than that of pure PLLA. This may be due to the plasticizing effect of PPD, where these blends are softer and tougher during elongation and form thin necks. However, the mechanical properties of the other blends 50/50 and 80/20 (PLLA/PPD) were not improved compared to the pure PLLA.
Ma et al. prepared a mixture of PLA with an amorphous material polypropylene carbonate (PPC), a degradable, aliphatic polycarbonate composed of different components. The tensile strength and modulus of all types of blends decreased with increasing PPC content. However, as the amount of PPC increases, the tensile toughness improves compared to pure PLA. The toughening effect is very obvious when the PPC content is higher than 40% or more. This is because PLA is a continuous phase when it is mixed with less than 30% PPC, while PPC becomes continuous when it is greater than 40% PPC. A continuous PPC phase facilitates the yielding of the substrate, i.e., more energy is required to break the material.
Liu et al. (2005) prepared a blend of PLLA with another biodegradable polyester, polyethylene terephthalate-tetramethylene ester (PTAT), by casting with chloroform solution and investigated its mechanical properties. It was found that the PLLA/PTAT blends exhibited interesting nonlinear stretching behavior at three fractions (75/25, 50/50 and 25/75 mass fractions of PLLA/PTAT). The tensile strength of the 75/25 PLLA/PTAT blend was 25 MPa and the elongation at break was 97%, while the tensile strength of pure PLLA was 28 MPa and the elongation at break was 19%. However, for the 50/50 PLLA/PTAT blends, the tensile strength and elongation at break were reduced to 7 MPa and 34%, respectively. This may be due to the poor compatibility and high phase separation of the blends. In addition, the tensile strength of the 75/25 PLLA/PTAT blend was slightly higher than that of the 50% PTAT (11 MPa), but the elongation at break was about 15 times higher than that of the pure PL LA (285%). These results suggest that PLLA is hard and brittle, while PTAT has higher plasticity.
Jiang et al. investigated the melt blending of poly(adipic acid-terephthalate-butylene glycol) (PBAT) with PLA. PBAT is a flexible, biodegradable aliphatic aromatic polyester with an elongation at break of 700%. If a certain amount (5%-20%) of PBAT is added to PLA, the tensile strength and modulus of the blends will be reduced. The tensile strength of pure PLA decreased from 63 MPa to 47 MPa with the addition of 20% PBAT, and the modulus of the blends (2.6 GPa) also decreased slightly when the PBAT content was 20 % compared with that of pure PLA (3.4 GPa). These results are predictable because PBAT has lower modulus and tensile strength than PLA. As the PBAT content increases from 5% to 20%, the impact strength of the cantilever beam increases and the best toughening effect is achieved when the PBAT content is 20%. The elongation at break increased significantly with the increase of PBA T content; even at 5% PBAT content, the elongation at break reached more than 200%. With the increase of PBAT content, the fracture mode changed from brittle fracture of pure PLA to ductile fracture of the mixture. SEM micrographs of the impact fracture surface have confirmed this, i.e., the increasing number and length of fibers on the fracture surface with increasing PBAT content. The SEM micrographs also indicate that the debonding-induced shear yielding mechanism is involved in the toughening of the blends.
Liu et al. reported the blending of PLA with different amounts of poly(ethylene/butylene succinate) Bionolle (Bignon) using a single-screw extruder. Bionolle is also a biodegradable aliphatic thermoplastic polyester. The elongation at break was slightly higher for different ratios of Beynen blends than for pure PLA, and the highest elongation at break was 8.2% at 40% of Beynen. However, the tensile strength and modulus of the blends decreased with the increase of the amount of piñon. This is due to the fact that the tensile strength and modulus of Bison are lower than those of PLA.
Shibata et al. reported the effect of polybutylene succinate (PBS) and polybutylbutyl-L-acid (PBSL) blended with PLLA. PBSL is a new type of PBS-based biodegradable polyester. Melt blending blends PPLA with PBS or PBSL and then injects it into the mold. The tensile strength and modulus of the blends generally decreased with increasing PBSL or PBS content, but the tensile strength and modulus of PLLA blends with 1% and 5% PBS were higher than those of pure PLLA. The authors concluded that this result can be attributed to the formation of finely dispersed co-elutions in the system, which was confirmed by field emission scanning electron micrographs. All blends exhibited high elongation at break over the entire range of components compared to pure PLLA, PBSL and PBS. Overall, the PLLA/PBSL blends exhibited higher elongation at break and lower tensile strength and modulus compared to the same percentage of PLLA/PBS blends.
Chen and Yoon compared the effects of adding untreated organoclay Cloisite 25A (hereafter C25A) and treated organoclay TFC on the mechanical properties of PLLA/polybutanedioic acid-adipic acid-butyrate (PBSA) composites. In this study, the composition mass ratio of PLLA/PBSA was fixed at 75/25 because the brittleness of PLLA was greatly improved in this mixture. Functionalized organoclay (TFC) was prepared by the reaction of glycine oxypropyltrimethoxysilane (GPS) with C25A. PLLA/PBSA/clay nanocomposites were prepared by melt blending PLLA and PBSA with organic clay at 180 °C. The researchers found that the tensile modulus of PLLA/PBSA composites with the addition of C25A and TFC was higher than that of PLLA/PBSA binary blends over the entire clay composition range. This is in line with expectations because the clay plays a reinforcing role in the composite. However, the elongation at break of the composites with the addition of organoclay C25A and TFC was much lower than that of the PLLA/PBSA blend. However, the elongation at break and modulus of the composites were improved for the treated clay (TFC) compared to the untreated clay (C25A). For example, the elongation at break for PLLA/PBSA/C25A with 10% C25A was 5.2%, while PLLA/PBSA/TFC with the same amount of TFC had an elongation at break of 46%. The tensile modulus and elongation at break of PLLA/PBSA/TFC composites were higher than those of PLLA/PBSA/C25A composites due to less agglomeration of the former than the latter. Therefore, this contributes to a higher degree of exfoliation and improves the interaction between the epoxy groups of TFC and the functional groups of PLLA/PBSA.
Chen et al. also reported similar work on mixing different amounts of untreated and treated clays with PLLA composites. Chen and Yoon reported that instead of just mixing PLI A and PBSA together, PLLA and PBS were blended with organic clay to improve the mechanical properties of the blends. Again, the mass ratio of PLLA/PBS was fixed at 75/25 and the same untreated organoclay C25A and treated organoclay TFC were used. The tensile modulus of PLLA/PBS composites with different amounts of C25A and TFC was higher compared to the PLLA/PBS blends. For example, when the composite contains 10% C25A organic clay, the modulus is 1.94 GPa, while a mixture without organic clay has a modulus of 1.08 GPa. This suggests that untreated and treated clays are used as reinforcing fillers because they have a high aspect ratio and flat structure. The tensile modulus values of the treated TFC-reinforced PLLA-PBS were more significantly affected by the clay content compared to the untreated C25A-reinforced PLLA/PBS. The modulus of PLLA/PBS blends containing 10% TFC was 1.99 Gpa.
However, the elongation at break of PLLA/PBS S composites decreased sharply after the addition of untreated C25A. Although in many cases compounding with clay decreases elongation at break, in contrast, the elongation at break of PLLA/PBS composites increases with increasing TFC content. The authors observed that composites containing treated clay (TFC) exhibited increased necking and significant fiber fracture surface formation, while composites containing untreated C25A exhibited brittle fracture without necking. This suggests that the chemical bonding between the TFC epoxy functional group and the two polymers of PLLA/PBS acts as a capacitor, thus increasing the interfacial interactions. The above studies are summarized in Table 5-6.
Table 5-6 Summary of mechanical properties reported for blends of PLA with degradable or partially degradable polymers
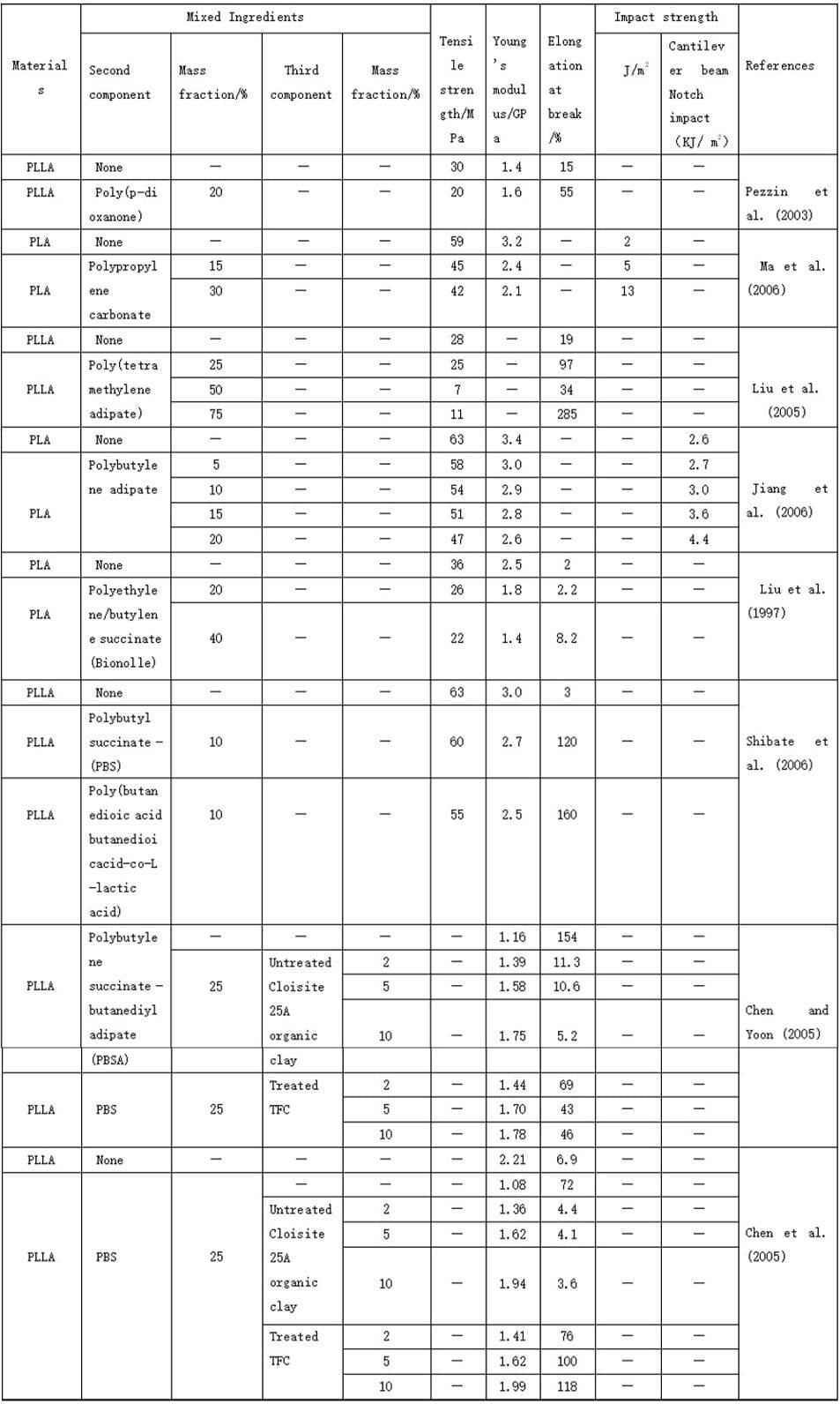
5.5.1 Mixtures of polylactic acid and polyhydroxy fatty acid esters
Polyhydroxyalkanoates (PHAs) are degradable linear polyesters produced in nature by a wide and common range of microorganisms. They are produced by bacteria and are used to store carbon and energy. Over 150 different monomers can be combined with this range to produce materials with very different properties. The best known types of PHAs include poly(3-hydroxybutyrate) (PHB) homopolymers, 3-hydroxybutyrate and 3-hydroxyvaleric acid (PHBV) copolymers, and poly(3-hydroxybutyrate)-co-(3-hydroxyalkanoate) copolymers. Since PHA is made from natural resources, the PHA/PLA blend is likely to be fully biodegradable. The mechanical properties of PHA/PLA blends have been reported by many researchers. iannac et al. reported a mixture of PLLA and PHBV prepared by casting with chloroform solution at room temperature. The elongation at break of the blends containing 20% and 40% PHBV increased slightly. However, the tensile strength and modulus of the blends decreased as the amount of PHBV was increased. This is due to the decrease in the crystallinity of the PLLA phase as the PH-BV content increases.
In a similar study conducted by Ferreira et al., it was found that the tensile strength of PLLA blends without the PHBV content was lower than the results of Iannace et al.. This is because the PLLA films obtained by Ferreira et al. were porous, unlike Iannace et al. who only obtained dense films. However, Ferreira et al. verified the trend of Young's modulus values of PLLA/PHBV blends found by Iannace et al.. PHB is the simplest and most common PHA. yoon et al. investigated the effect of different types and amounts of bulking agents on the mechanical properties of PLLA/PHB blends. PLLA and PHB (50/50 by mass) were mixed in trichloromethane (3%) and then the films of PLLA/PHB mixture were recovered by evaporating the solvent and drying in vacuum at 40 °C. The bulking agents used were PLLA-PEG-PLLA triblock copolymer, PEG-PLLA diblock copolymer, and 2% and 5% polyvinyl acetate (PVAc). The elongation at break and tensile toughness of all the blends with the addition of capacitators (2% and 5%) were improved compared to the PLLA/PHB blends without capacitators. However, the tensile modulus of the blends with different amounts of compatibilizers was reduced compared to the PLLA/PHB blends without compatibilizers. The tensile strength results varied with the type and composition of the bulking agent. The highest tensile strength was obtained for the blend prepared with 2% PLLA-PEG-PLA triblock copolymer (69.8 MPa), followed by 2% PEG-PLA diblock copolymer (65.5 MPa), while the tensile strength of the blend prepared with 5% diblock copolymer and triblock copolymer and PVAc as compatibilizer was lower than that of the PLLA/PHB without solubilizer blends without solubilizers.
From the perspective of tensile strength, elongation at break and toughness, PLLA/PHB blends with 2% added PLLA-PEG-PLA triblock copolymers are the best formulation choice, and despite a slightly lower Young's modulus, the mechanical properties of PLLA/PHB blends are higher than those of PLLA/PHE blends without solubilizers.
Takagi et al. prepared blends of PLA and the biodegradable thermoplastic polymer poly(3-hydroxy fatty acid) (PHA) in different compositions. PLA was blended with functionalized PHA (ePHA) containing 30% epoxy group in the side chain. They found that the impact strength of the simply supported beam (Charpy) of both PLA blends increased with the addition of either PHA or ePHA. These results were higher than those of pure PLA, but the tensile strengths of both PLA/PHA and PLA/ePHA blends were lower than those of pure PLA. the impact and tensile strengths of the PLA/ePHA blends were higher than those of the PLA/PHA blends compared to the two blends. This is because the addition of epoxy side groups of ePHA improved the compatibility of the blends.
Noda et al. prepared a blend of PLA with biodegradable PHA by melt blending with a single-screw extruder. The PHA used is a poly(3-hydroxybutyrate)-co-(3-hydroxyalkanoate) copolymer called Nodax developed by Procter & Gamble. The authors found that the addition of 10% Nodax significantly improved the toughness of the copolymer. They found that the tensile energy at break, calculated from the area under the tensile stress-strain curve, was 10 times higher than that of pure PLA. However, it is noteworthy that this effect is only observed for Nodax up to 20%. In fact, further incorporation of Nodax will reduce the toughness of the blend to the original level of pure PLA. This is because when Nodax is below 20%, the copolymer is delicately dispersed in the PLA matrix as a discrete phase, which means that the PHA portion of the blend remains mainly in the liquid amorphous state, thus preventing crystallization. The reduced crystallinity makes the blend ductile and tough.
Schreck and Hillmyer reported similar work on the melt blending of Nodax with PLLA. The Nodax composition varied from 0% to 25% using a Haake intermittent mixer at 190°C and 75 r/min for 15 min. Rather than studying the tensile energy at fracture as reported by Noda et al. (2004), Schreck and Hillmyer investigated the effect of PLLA/ Nodax blends on the notched impact strength. A similar improvement in toughness was observed in blends up to 20% Nodax. The highest impact strength was obtained with blends containing 15% Nodax, with a value of 44 J/m, compared to 22 J/m for pure PLLA. To improve the performance of the binary blends, Schreck and Hillmyer also investigated the effect of the terpolymer PLLA/Nodax and oligoNodax-b-poly(L-propyleneglycol) diblock copolymers as bulking agents. The amount of oligomeric Nodaxb-poly(L-propyleneglyceride) was fixed at 5% in the mixture with a mass ratio of 81/14PLLA/Nodax. However, the addition of 5% oligomeric Nodax-b-poly(L-propyleneglyceride) did not show any improvement in toughness. This is due to the poor interfacial bonding due to the low degree of oligomer-polymer entanglement at the particle matrix interface, which reduces the deformability and the ability to dissipate impact loads. A summary of the literature on PLA and PHA blends is presented in Table 5-7.
Table 5-7Mechanical properties of poly(lactic acid)/polyhydroxyalkanoate (PHA) blends
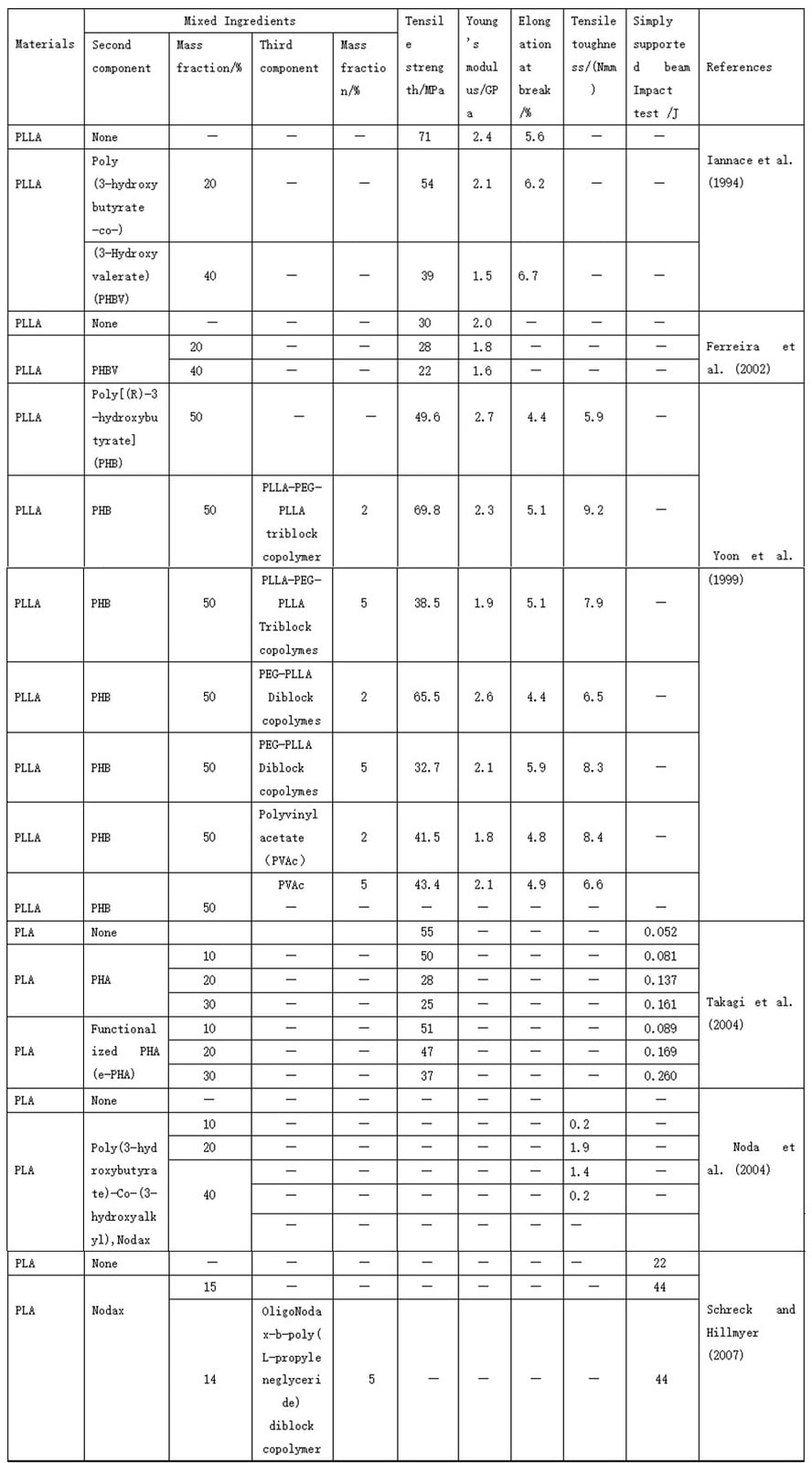
5.5.2 Blends of PLA with non-degradable polymers
The blending of PLA with non-degradable polymers has not been as widely studied as the blending with degradable or renewable resource polymers. However, blending PLA with commercial polymers can be very useful, especially in improving processing properties, reducing costs and controlling biodegradation rates. Kim et al. (2001) investigated the effect of blending high molecular weight PEO with PLLA. The blend fraction of PLLA/PEO in the blend was fixed at 60/40. In addition to the PLLA/PEO blend, they added PVAc as a bulking agent at different concentrations (2% to 20%) and prepared the blends by solution and melt blending. Solution blends were made in trichloromethane at 3% while melt blends were prepared using a Brabender (PlastiCorder) mixer. The researchers found that the tensile strength of the solution blends was higher than that of the melt blends at the same PVAc loading. However, the elongation at break of the solution blends was lower than that of the corresponding amount of PVAc melt blends.
The addition of different amounts of PVAc to the solution blends resulted in a slight decrease in tensile strength and an increase in elongation at break. When 2% of PVAc was added, the elongation at break was significantly increased, but the tensile strength was not significantly decreased. Meanwhile, the tensile strength of PEO/ PLLA blends increased with the increase of PVAc usage. Elongation at break continues to increase until the addition reaches 5%, after which continued addition of PVAc results in a significant decrease in tensile strength. However, Kim et al. (2001) did not have any morphological data to confirm the difference between melt and solution blends.
Jim et al. investigated the effect of blending different amounts of polyisoprene with PLA. They found that the addition of 20% polyisoprene resulted in reduced elongation at break and tensile toughness relative to pure PLA. However, when PLA was blended with polyisoprene/PVAc graft copolymer, its elongation at break and tensile toughness were slightly increased compared to pure PLA.
Li et al. reported the effect of blending PLA with organically modified montmorillonite head (MMT) nanoclay (Cloisite 30B) and blending PLA/Cloisite 30B with core (polybutyl) shell (polymethyl methacrylate) rubber (Paraloid EXL2330). The MMT nanoclay surface was modified by ion-exchange reaction followed by cationic modification with bis(2-hydroxyethyl)methyl(hydroxylated alkyl)ammonium. The addition of 5% Cloisite 30B improved the modulus of the blends but reduced the tensile strength and elongation at break. However, the blend of PLA/Cloisite 30B (5%) and Paraloid EXL2330 (20%) significantly increased impact strength (134%) and increased elongation at break (6%) compared to pure PLA, while modulus remained almost constant and only decreased tensile strength (28%).
NatureWorks, the largest manufacturer of PLA, reports the use of a high rubber content (35% to 80%) impact modifier, Blendex 338, an acrylonitrile-butadiene-styrene terpolymer containing 70% butadiene rubber, to improve impact strength. The addition of 20% Blendex 338 to the PLA blend improves notched impact strength and elongation at break. The notched impact strength increased from 26.7 J/m to 518 J/m and the elongation at break increased from 10% to 281%. NatureWorks (2011) also reported the toughening effect of mixing PLA with a polyurethane provided by Dow Chemical (i.e., Pellethane 2102-75A) on PLA. The addition of 30% Pellethane 2102-75A increased notch impact strength from 26.7 J/m to 769 J/m and elongation at break from 10% to 410% significantly.
DuPont developed Biomax Strong, a petroleum-based impact modifier for modifying PLA (Dupont). It is an ethylene copolymer that improves the toughness and reduces the brittleness of PLA materials. biomax Strong can still reduce the brittleness of PLA at levels as low as 2%. It also enhances the impact strength, flexibility and melt stability of PLA; these properties are particularly useful in rigid applications, such as cast sheets for thermoforming and injection molding. When used at the recommended 15%, Biomax Strong outperforms competitive products in terms of improved toughness with minimal impact on clarity. This product provides good contact clarity when used at recommended amounts and provides a more transparent container than other alternatives. By using the recommended amount of Biomax Strong, the performance of PLA is enhanced and compostability requirements are still met.
Anderson and Hillmyer investigated the blends of PLLA with linear low density polyethylene (LLDPE) (PLLA/ LLDPE) and high density polyethylene (HDPE) (PLLA/HDPE). In addition, they compared the effect of combining copolymer poly(L-propylene-co-poly(PLLA-PE) with PLLA/LLDPE co-blends and copolymer poly(L-propylene-c0-poly(ethylene-propylene-propylene) with PLLA/HDPE blends Compared to 20 J/m for pure PLLA, the addition of 20% LLDPE to PLLA resulted in a significant increase in impact strength significantly to 490 J/m. The impact value was further increased to 760 J/m by adding 5% of PLLA-b-PE block copolymer to the blended system. The elongation at break of PLLA binary and ternary blends was increased to 23% and 31%, respectively, and the elongation at break of pure PLLA was increased to 4%. However, both PLLA/LLDPE blends and their copolymers reduce the tensile strength and modulus of the material. When comparing PLLA with LLDPE and HDPE blends, it is found that when PLLA is blended with HDPE (even copolymers of HDPE), its tensile strength and modulus are lower than those of LLDPE blends. This is because the discrete rubber phase of LLDPE gives more adhesion to the PLLA matrix compared to the rigid HDPE, thus increasing the degree of toughening.
A recent study by Balakrishnan et al. also focused on blending PLA with LLDPE; however, they did not add only LLDPE, but also organically modified montmorillonite (MMT). The composition of LLDPE was fixed at about 10%, and the content of MMT varied from 2 to 4 parts by mass. The researchers found that as the MMT content in PLA/LLDPE blends increased, Young's modulus and flexural modulus increased and tensile and flexural strength decreased. This suggests that MMT is effective in improving the stiffness of the blends. It was demonstrated by transmission electron microscopy and X-ray diffraction that the component spacing of MMT in the blends increased to form intercalated nanocomposite systems (Balakrishnan et al,). Well-dispersed MMT lamellae contribute to the enhancement of PLA nanocomposites toughened by LLDPE.
Jiang et al. compared the mechanical properties of PLA nanocomposites prepared by nanoprecipitated calcium carbonate (NPCC) and organically modified MMT. PLA nanocomposites were prepared by melt blending with different mass fractions of NPCC and MMT (2.5%, 5% and 7.5%) using an isotropic twin-screw extruder. They observed that the elongation at break increased only when the MMT content increased to 2.5%, and the elongation at break increased as the NPCC content increased from 2.5% to 7.5%. The tensile strength of PLA nanocomposites decreases with increasing NPCC content and increases with increasing MMT up to 5%. Meanwhile, the Young's modulus of PLA increased slightly with the addition of NPCC and increased significantly with the addition of MMT. The results show that both nanoparticles have good dispersion at a filling amount of about 5%. However, as the number of nanoparticles increases, large agglomerates will be observed. The authors concluded that the different enhancement effect of the two nanoparticles may be mainly attributed to the different microscopic see structures of the nanocomposites and the interaction between the nanoparticles and PLA. Table 5-8 summarize the mechanical properties of PLA blends with non-degradable polymers.
Table 5-8 Mechanical properties of PLA and non-degradable polymers
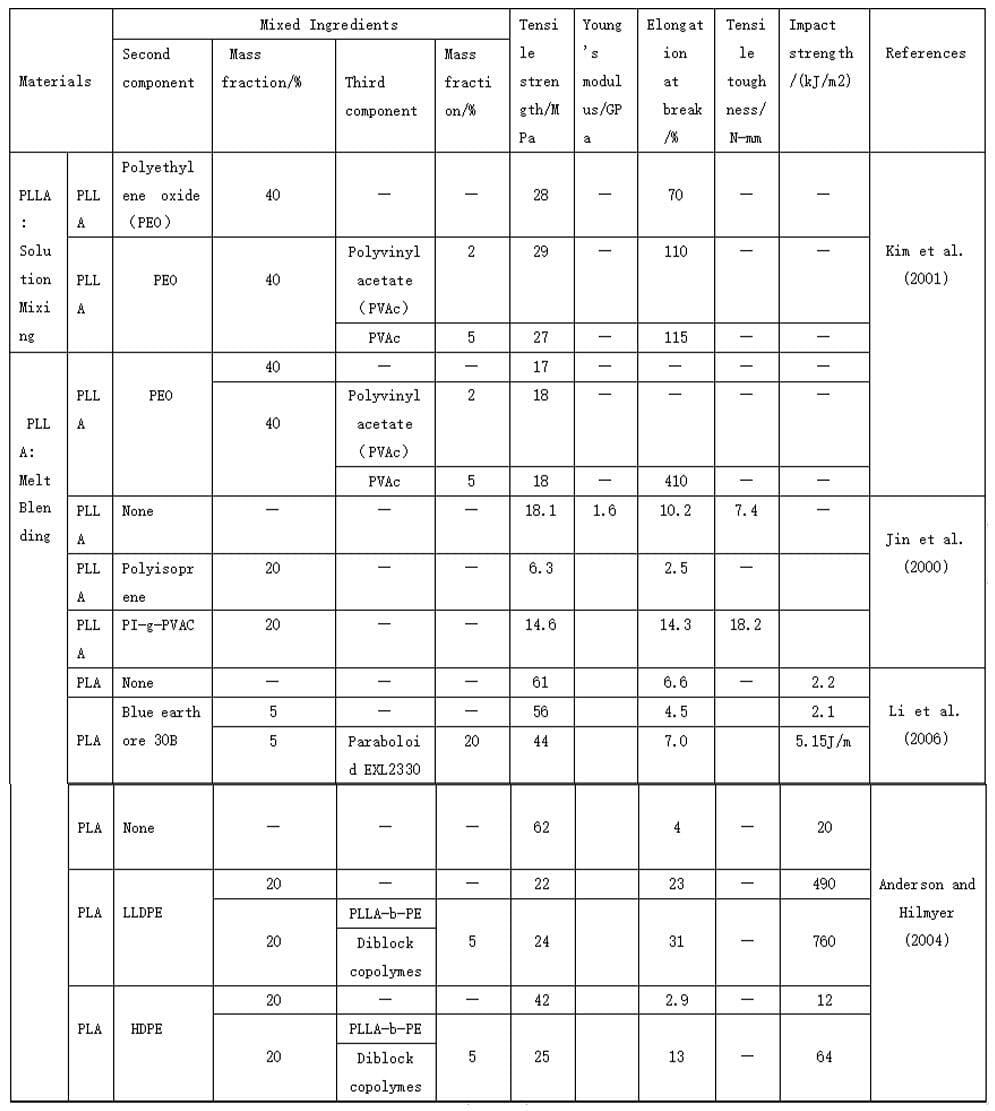
5.6 Conclusion
The mechanical properties of PLA can be changed by altering its stereo-configuration, crystallinity, molecular weight, etc. PLA with high stereoconfiguration purity has high tensile strength and high modulus, but its impact strength is low. In contrast, the copolymers of L-propyl cross-ester and D-cross-ester are still in amorphous form and have poor mechanical properties. Researchers tend to use copolymerization techniques to modify the existing properties of PLA to broaden its applications. In addition, polymer blending techniques have been used to combine the properties of PLA with those of another polymer to obtain better impact and flexural strength. In general, most modifications to PLA are aimed at improving its mechanical properties while maintaining its biodegradability. It is likely that this trend in PLA will continue in the coming decades.
6. Rheological properties of polylactic acid
6.1 Introduction
The definition of rheology is the study of the deformation and flow of fluids. It is an important property of molten polymers that relates viscosity to temperature and shear rate, and therefore to the processing properties of the polymer. Most polymer melts are classified as shear thinning fluids, i.e., at higher shear rates, the polymer molecules are oriented and the number of entanglements between polymer chains is reduced. These phenomena contribute to the easier passage of polymer molecular chains through each other into narrow cavities during polymer forming. The kinetic energy of the molecules is also higher at higher temperatures, which reduces the polymer viscosity.
Rotational rheometers and capillary rheometers are devices that can be used to obtain polymer shear viscosity data. Rotational rheometers are mainly used for low shear rate analysis from 0.001 to 100s-1. These instruments typically consist of a cone and a flat plate geometry; the cone angle is designed to maintain a stable shear rate during analysis. Capillary rheometers drive the melt flow by pressure and measure the pressure at the inlet of the capillary port mold to obtain apparent rheological data of the polymer melt. These data are displayed as viscosity and shear rate values, which are further corrected using Bagley and Weissenberg-Rabinowitsch correlations. Capillary rheometers are used to measure shear rates from medium to high, from 10 to 10,000 s-1. This measurement requires more material and time than the rotational method.
Polylactic acid (PLA) is made into useful articles by thermal processing, such as injection molding and extrusion. Therefore, its rheological properties, especially shear viscosity, have an important influence on thermal processes such as blown film, coated paper, injection molding, sheet molding, and fiber spinning. Therefore, it is important to study the rheological properties of PLA to understand the processing performance of PLA materials.
6.2 Rheological properties of polylactic acid
The melt rheology of PLA has a profound effect on the flow state of the polymer during processing. In general, the melt viscosity of high molecular weight PLA is 500-1000 Pa · s at a shear rate of 10-50 s-1. This polymer grade corresponds to a molecular weight (Mw of about 100,000) for injection molding and a molecular weight (300,000) for film casting extrusion applications (Garlotta). The melt of high molecular weight PLA behaves as a pseudoplastic non-Newtonian fluid, while the melt of low molecular weight PLA (40,000) exhibits Newtonian-like properties during a typical film extrusion process. Under the same processing conditions, semi-crystalline PLA has higher shear viscosity than amorphous PLA. In addition, the viscosity of the melt decreases significantly with increasing shear rate, i.e., the polymer melt exhibits a shear thinning behavior. This phenomenon was discovered by Fang and Hanna, who analyzed it using a tube rheometer mounted on an extruder. Its study analyzed the viscosity of two PLA resins (amorphous and semi-crystalline forms) at 150°C and 170°C. The viscosity data were calculated from pressure distribution and volume flow rate as a function of resin type, temperature and shear rate. The results in Figure 6-1 show that semi-crystalline PLA has higher viscosity than amorphous PLA at high temperature due to the difference in molecular structure. This is because the molecular structures are different. The molecules of semi-crystalline PLA are arranged in an orderly manner, with strong intermolecular forces and high resistance to flow. In contrast, the arrangement of molecules in amorphous PLA is random, which in turn exhibits less resistance to flow. In general, crystalline materials have better mechanical and mechanical properties than amorphous materials.
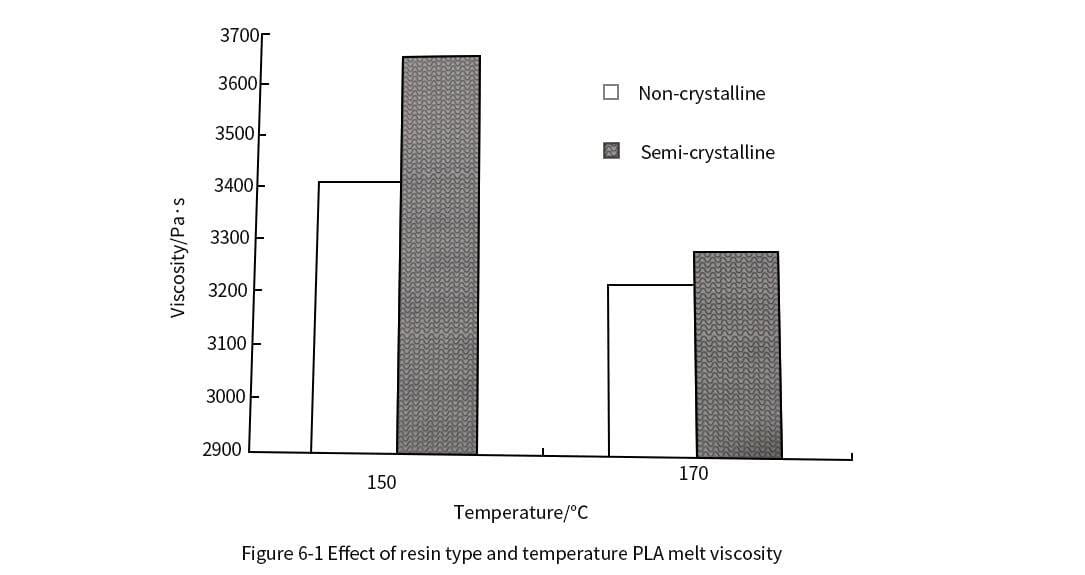
The increase in temperature leads to a decrease in the shear viscosity of both semi-crystalline and amorphous PLA. It can be seen that the viscosity values at 150°C are significantly higher than those at 170°C. This can be explained by the fact that at high temperatures, the connections between the molecular chains become weaker due to the high amplitude of the PLA molecules, allowing for a smoother melt flow. In addition, the shear rate has a significant effect on the viscosity of the PLA melt. As shown in Figure 6-2, η decreases sharply as the shear rate of both types of PLA increases. The relationship between η and shear rate is nonlinear, but shows a typical non-Newtonian pseudoplastic row force. This is mainly due to the breakage of molecular chains by the strong shearing action during extrusion.
The work of Fang and Hanna also summarized the power-law equations for amorphous PLA and semicrystalline PLA,from rheological tests using a single-screw Brabender extruder with L:D and compression ratios of 20:1 and 3:1, respectively. A nonlinear regression analysis of the power-law equations revealed that the correlation coefficients (r²) of all equations were greater than 0. 99 and the mean square misunderstanding of the power-law equations was small. This further demonstrates that both amorphous and semi-crystalline PLAs exhibit typical non-Newtonian pseudoplastic behavior. Nonetheless, NatureWorks' test results using Ingeo injection grade materials tested by capillary variometry showed a good match to the Cross-Williams-Landel-Ferry equation (WLF) viscosity model. There are seven coefficients in the model that can be easily embedded into Moldflow injection molding simulation software (Moldflow Plastic Labs). Moldflow is a computer software widely used in the plastic injection molding industry to predict and optimize the injection molding process, and to assist in mold design. The Cross-WLF model provides insight into the effects of various injection molding conditions, such as pressure and temperature as the polymer melt flows into a narrow mold cavity.
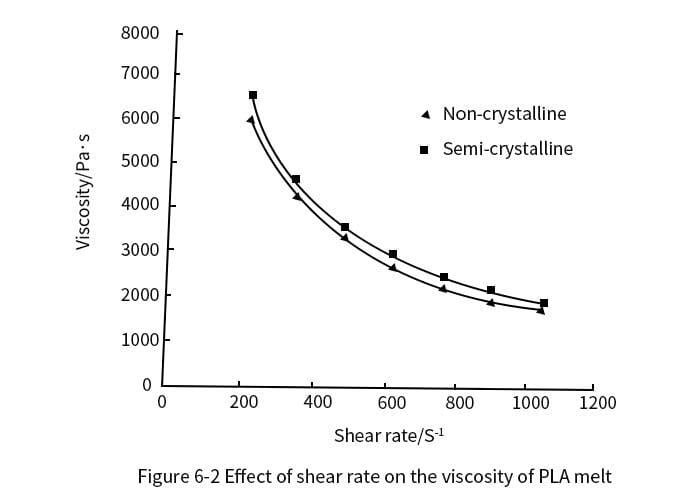
6.3 Effect of molecular weight
The viscoelastic properties of PLA melts of different molecular weights were measured over a range of temperatures, frequencies and shear rates using parallel plate geometries. Cooper-White and Mackay performed a typical study using a Rheometrics RDSII torque rheometer with 7.9 mm stainless steel parallel plates to test three commercial grade poly(L-lactic acid) (PLLA) with significantly different molecular weights (40,000, 130,000 and 360,000 Mw, respectively). tests.
The viscosities of PLLA melts were compared with those of conventional polymer melts using an empirical formula for the zero-shear viscosity (ηo) versus molecular weight of linear amorphous polymers. The form of this equation has been applied to the elasticity coefficient AG to further quantify the effect of molecular weight on elasticity (Cooper White, Mackay, 1999):
η0 = K(M w)a (6-1)
A G =K′(M W)b (6-2)
In these equations, the constants K and K′ depend on the polymer type, molecular weight and temperature. As shown in Figure 6-3, the power law factors "α" and "b" are obtained from the slope of the log viscosity coefficient VS log molecular weight curve and the slope of the log elasticity coefficient VS log molecular weight curve, respectively. Theoretically, the molecular weight index "α" is predicted to have a universal value of 3.4 greater than Mc (the critical molecular weight of the entangled state of a linear flexible polymer) (Piver). Many systems follow this relationship. The elasticity coefficient of monodisperse polystyrene as a function of molecular weight, expressed by the exponent "b", has been shown experimentally to be around 7.5 (Onogi et al.).
From Figure 6-3, the following equation is obtained:
η0 = (3×10-17Pa·s)(M w)4.0 (6-3)
A G = (2×10-38Pa·s2 )(M w)8.0 (6-4)
The value of the molecular weight index relative to zero shear viscosity is slightly higher than the generally accepted value of 3.4. The elasticity coefficient of PLLA melts is more dependent on molecular weight at 8.0 compared to monodisperse polystyrene melts. This deviation is thought to be caused by spatial site resistance-overwinding expansion due to possible differences in chemical shifts within and between PLLA polymer chains (including tertiary chain-chain interactions).
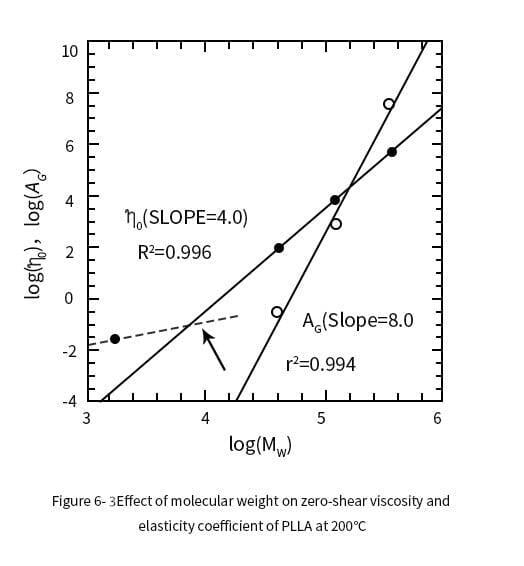
Witkze showed that the effect of temperature on η0 for 15% D-propyl cross ester PLA can be described as (Witzke):
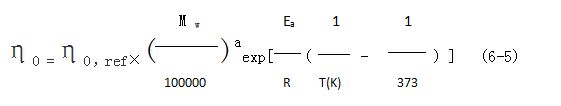
where α=3.38±0.13, fluid activation energy Eα=190kJ/mol, η0,ref= 89400±9300Pa-s,R is the gas constant="8.314J/(k-mol),T" is the temperature (K). The zero shear viscosity η0 can be related to the isomeric components by fitting the known WLF equation (Witzke).
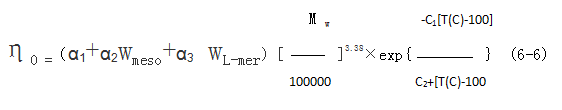
where, Wmeso and WL-mer are the initial mass fractions of racemic propyleneglyceride and L-propyleneglyceride, respectively, α1=one 3000, α2=-42000, α2=112000, C1="15.6±1.6" and C2="(110±11)°C;" α1, α2, α2 and C1 have no units; T(C) test temperature unit is °C.
Equation (6-6) can be used to predict η0 for amorphous PLA containing more than 50% of the L-monomer fraction between Tg and Tg + 100 °C. This equation predicts that η0 increases with increasing L-monomer content and decreases with increasing racemic propanediol content.
6.4 Impact of Branching
The introduction of chain extenders can significantly improve the rheological properties of PLA. Since linear polymers exhibit low melt strength in some applications, it is desirable to improve the melt strength by introducing long chain branches. There are several ways to promote the branching of PLA, such as the use of multifunctional polymerization initiators, hydroxycyclic ester initiators, polycyclic esters and crosslinking via radical addition (Leher meier, Dorgan).
6.5 Tensile viscosity
Tensile viscosity studies on high L content PLA (Mw = 111000 to 120,000) have shown that PLA can be stretched to large strains without fracture. Polymers also exhibit strain-hardening behavior during deformation (Palade et al.), which is an important feature in processing operations such as fiber spinning, film casting, and blown film. This tensile measurement was performed on a Rheometrics tensile rheometer using rectangular samples at 180°C and a stretch rate of 0.1s-1. The most striking feature of the response is the occurrence of a very strong strain hardening (two orders of magnitude increase in tensile viscosity). This effect is most pronounced when long branched chains are present, as it makes the relaxation time longer.
6.6 Solution viscosity of PLA
Although the solution viscosity of PLA in a solvent is not directly related to the processing of molten PLA polymers, this property is usually evaluated for quality control purposes to determine the molecular weight of the resin and the processed part. The relationship between viscosity and the molecular weight of PLA dissolved in dilute solution is usually modeled using the Mark-Houwink equation:
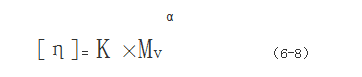
where [η] is the characteristic viscosity, K and α are constants, and Mv is the experimental viscosity-averaged molecular weight. the Mark-Houwink equation depends on the type of PLA, the solvent used, and the temperature of the solution.
6.7 Rheological properties of polymer blends
Polymer blending techniques can improve the properties of PLA. To improve the properties of PLA and to obtain new materials, PLA has been blended with several synthetic polymers and biopolymers. PLA has been blended with rubber, thermoplastic starch, polybutylene succinate, polybutylene succinate, polybutylene adipate-terephthalate (PBAT), acrylonitrile-butadiene-styrene, polypropylene, polyethylene, polystyrene, and laminated silicates to obtain lower-cost, better-performing materials.
6.7.1 Poly(lactic acid)/poly(butylene terephthalate) adipate blends
The steady-state shear rheological behavior of PLA/PBAT blended melts at different PBAT blending ratios exhibits a typical non-Newtonian fluid (Gu et al.).
A study by Gu et al. used a twin-screw extruder with a screw diameter of 27 mm and an L/D ratio of 42 to prepare PLA/PBAT blends by melt blending. The degradation of PLA during heating was eliminated by using about 0.5% (mass ratio of PLA/PBAT) of TNPP as a stabilizer, which acted as a chain extender, rejoining the polymer chains broken by humidity and temperature increase.
The data were fitted using power law equations, and the results showed that the correlation coefficient (r2) was greater than 0.99 for all equations. The addition of PBAT leads to a decrease in the flow index n. The temperature dependence of polymer melt viscosity is one of the most important parameters in polymer flow. In a certain temperature range, the dependence can be expressed in Arrhenius form:
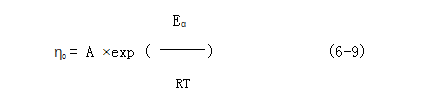
where η0 is the zero shear viscosity, R is the gas constant, A is a constant, and Eα is the flow activation energy. A higher Eα will result in a melt that is more sensitive to temperature changes. The Eα values at 160°C, 170°C and 180°C were calculated using η0 data. The results are very consistent with the Arrhenius model, and it is clear that Eα decreases with the addition of PBAT. The low temperature dependence of PLA/PBAT melts simplifies the selection of processing temperatures for the blends. That is, PLA/ PBAT blends have a wide processing temperature window due to their low viscosity sensitivity to temperature.
6.7.2 PLA/Polystyrene blends
Hamad et al. investigated the blending of petroleum-based polystyrene with PLA to improve the stiffness of the blends and to determine their rheological behavior at high shear rates. The mechanical properties of PLA/Polystyrene are not discussed in this section. However, the rheological properties of PLA/polystyrene blends were investigated using a Davenport 3/80 capillary rheometer at 165°C, 175°C, 185°C and 195°C and at capillary conditions of L/R=8, 15, 25 and 36. The blends were prepared using a laboratory-scale single-screw extruder at 30%, 50%, and 70% (mass fraction) polystyrene ratios, abbreviated as PLA70, PLA50, and PLA30, respectively (Hamad et al., 2010). The rheological curves of these blends at 165 °C are shown in Figure 6-4.
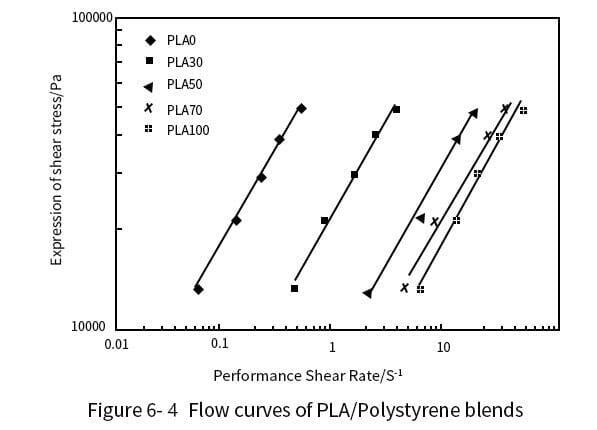
It can be seen that the linearity of these lines is high and they obey a power law for a certain range of shear rates. The value of the power law exponent n calculated from the slope of the fitted straight line is less than 1. This means that PLA/polystyrene blends melts are pseudoplastic fluids, similar to most thermoplastic polymer melts. Figure 6-5 shows the plot of true viscosity versus true shear rate for the PLA/polystyrene blends at 165°C. PLA polystyrene and its blends exhibit a typical shear thinning behavior in the studied shear rate range. This behavior may be due to the alignment of the polymer chain segments in the direction of the applied shear stress.
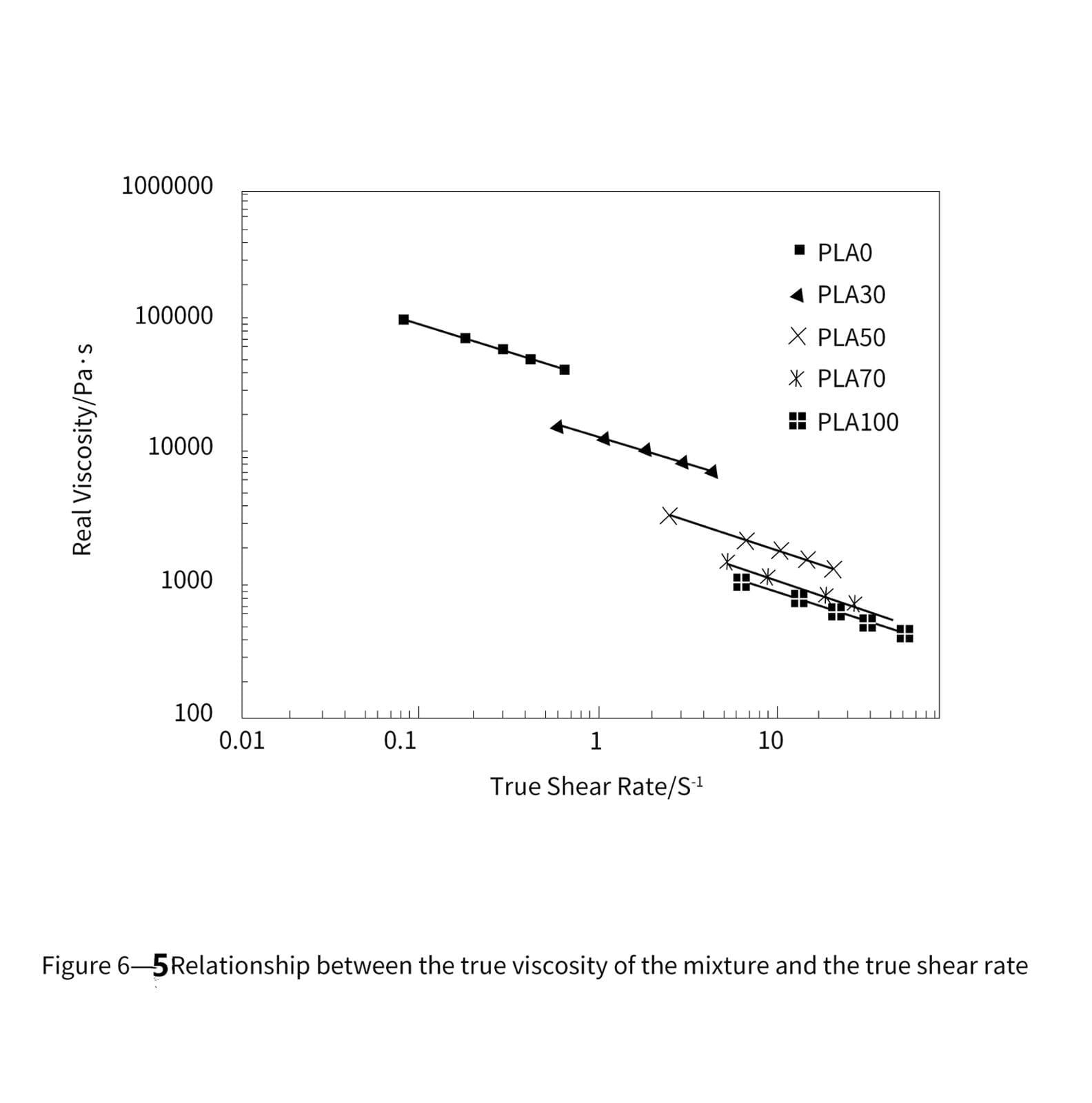
The curves of the true viscosity and PLA content of the mixture for shear rates of 10s-1 and 100s-1 are shown in Figure 6-6.
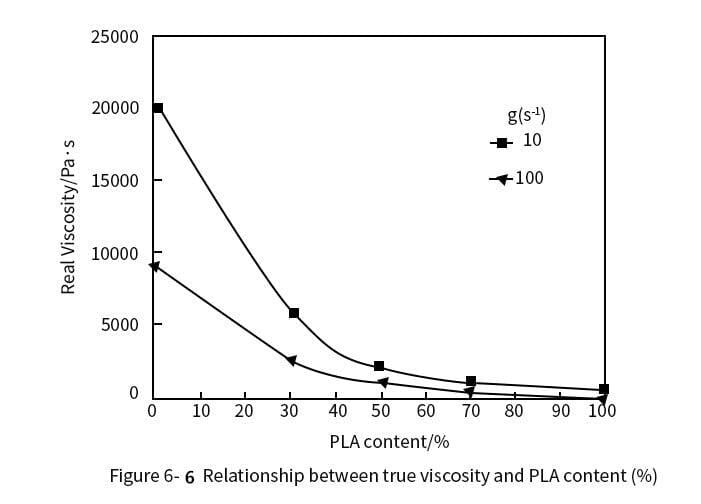
The results showed that the viscosity of polystyrene was higher than that of pure PLA, and the viscosity of the blends increased with the increase of polystyrene content. This effect is clearly visible as the polystyrene content increases. This phenomenon is caused by the inherent high viscosity of polystyrene. These results are important because they suggest that the optimal process conditions for molding and processing PLA/polystyrene blends may be very different compared to pure PLA. The addition of 30% PLA to polystyrene resulted in a 0.7-fold decrease in the true viscosity (ý =10s-1), which may be caused by the poor compatibility of PLA with polystyrene.
The activation energy of flow at constant shear stress (Eτ) and constant shear rate (Eý) can be obtained from the slope of the graph as follows:

It is well known that the magnitude of the flow activation energy reflects the sensitivity of the viscosity to temperature, therefore, the higher the Eτ or Eý, the higher the sensitivity of the blend to temperature. From the values of Eτ and Eγ, it can be seen that Eτ increases with increasing shear stress, while Eγ decreases with increasing shear stress. It can also be seen that Eτ > Eγ and Eγ/Eτ < 1, confirming that PLA70 is a pseudoplastic material (Han).
6.8 Conclusion
The rheological properties of PLA in its amorphous and semi-crystalline state have an important influence on the processing properties. Although different types of PLA and their blends have been studied, many results indicate that PLA maintains non-Newtonian pseudoplastic behavior under high shear conditions. Various models have been developed to describe the rheological behavior of PLA and its blends. These models are important for predicting the processing behavior and revealing the molecular interactions under shear. Due to the expanding applications of PLA, it is believed that improvements in the rheological and processing properties of PLA will contribute to the future development of polymer production technology.
7. Degradation and stability of polylactic acid
7.1 Introduction
As described in Chapter 6 on the rheological properties of PLA, polylactic acid or PLA is known for its environmental qualities and is much "greener" than commodity plastics such as polyethylene, polypropylene, polystyrene and polyvinyl acetate in packaging applications. While several aliphatic polyesters including poly(lactide) (PCL), polyhydroxy fatty acid esters (PHA) and polybutylene succinate (PBS) are also biodegradable, PLA has the advantage of being produced by lactic acid fermentation of sugars, allowing for large-scale production. Although PCL and PBS are also biodegradable polymers, they are produced from petrochemical resources. This further highlights the advantages of PLA, whose production reduces greenhouse gas emissions. However, further development of PLA is needed in order to increase yields.
In order to meet current stringent environmental regulations, it is critical to understand the biodegradability of PLA and the role of biodegradation for the plastics industry. In addition, PLA and its copolymers have been used in medicine for decades, so it is essential to evaluate and control their biodegradability in living organisms. Currently, most of the PLAs on the market are synthesized by ring-opening polymerization of the cyclic dimer of lactic acid, propylene glycol. Lactic acid has two stereoisomers, D and L, whose composition has an important influence on the mechanical properties and biodegradability of PLA. L-lactic acid is produced naturally by the fermentation of microorganisms. Small amounts of D-lactate are produced incidentally by some bacterial species. Chemical synthesis has contributed significantly to the production of PLA. Since D-lactic acid does not occur in large quantities in nature, cells and microorganisms lack the ability to metabolize this lactic acid or have a very low capacity to do so. Most of the degradation of D-lactate is through hydrolysis reactions that convert it into simple molecules. After polymerization, the stereomorphic forms of PLA-poly(L-lactic acid) (PLLA), poly(D-lactic acid) (PDLA), and poly(D,L-lactic acid) (PDLLA)-show different melting points and crystallinity depending on the isomeric composition of PLA. In the development of medical PLA, controlling the average molecular weight (Mw) of PLA plays an important role in controlling the corrosion of PLA. The addition of the D-propyl cross-ester isomer also helps to reduce the degradability of PLA in body fluids and tissues. This is because mammals cannot produce a suitable enzyme to act on D-lactate. Thus, hydrolysis reactions are thought to be involved in the reduction of D-lactate prior to its assimilation by the liver. Although PLA has been known and used for decades, information on its degradation and consumption by microorganisms and living tissues is still limited (Tokiwa, Calabia). This chapter provides an overview of the degradation of PLA by microbial, enzymatic and living tissue action, as well as by thermal radiation and incineration.
7.2 Factors affecting the degradation of PLA
The degradation of polymers is mainly caused by the erosion of external elements. This is due to the fact that polymer chains are highly stable and rarely undergo autocatalytic reactions. Although PLA is produced from lactic acid produced by organic processes (bacterial fermentation of sugars), its conversion to PLA leads to significant changes in biological and chemical degradation mechanisms. PLA cannot be broken down and consumed directly by living cells as efficiently as lactic acid itself. Stereochemistry, crystallinity and molecular weight are the main factors affecting the biodegradation behavior of PLA.
First, to better understand the degradation mechanism, several terms need to be clarified. The definitions of these terms are summarized in Table 7-1. It is important to distinguish between "biodegradation" that occurs in the environment and "biodegradation" that occurs in living tissue. This is because biodegradation in the environment is caused by the action of microorganisms, while biodegradation in tissues refers to degradation processes that occur in vivo or in vitro and are associated with reactions of enzymes and components in cells or body fluids. PLA is a biopolymer that has a wide range of applications in both everyday life and biomedical fields. It can be used as packaging material, as well as surgical sutures and implants.
Table 7-1 Definitions of Common Biological Terms for Biopolymers
①General terminology from British Standard Institutions BS EN13432.
② from the generalized terminology of Woodruff and Hutmacher.
In general, the degradation of polymers is essentially influenced by chemical bonding. The low-reactive backbone of polymers is virtually invulnerable to external elements. This is obvious for polymers that do not contain negatively charged elements (especially oxygen), as they are less likely to be hydrolyzed and therefore can remain unchanged for longer periods of time. In his review article, Göpferich compares a number of hydrolyzable polymers, poly(anhydride), poly(protic acid) esters and poly(koolaid). He found that the oxygen-containing backbones of poly(anhydride) and poly(protic acid) esters are most susceptible to hydrolysis reactions.PLA belongs to the polyester class and takes a long time to reach its hydrolysis half-life. This is due to spatial effects:A large number of alkyl groups hinder water intrusion (Göpferich). Polyvinyl alcohol (PVOH) is another hydrolysable polymer. PVOH has a high rate of hydrolysis in the presence of hydroxyl groups (OH) because hydroxyl groups protrude and form hydrogen bonds easily. Although PLA is a polar polymer similar to PVOH, it lacks water-sensitive behavior as a hydrophobic polymer. Therefore, in order to initiate hydrolysis processes related to microbial or tissue/organ assimilation, PLA needs to be exposed to water for a long period of time.
The composition of the copolymer also affects the degradation of the polymer. Table 7-2 shows the approximate degradation times of pure polymers and their derived copolymers. One of the main reasons for the change in degradation kinetics of copolymers is that the additional monomers affect the crystallinity and reduce the spatial effect (Hiemenz). It was found that the rate of chain breakage increased with the increase of ethyl cross ester content. Conversely, copolymerization of L-propyl cross-ester with D,L-propyl cross-ester increased the degradation time. This is due to the fact that D-lactic acid is unlikely to be naturally degraded by the body's enzymes. This approach helps to extend the time that PLA implants can function in the body. PCL is a biopolymer that can be consumed by bacteria and fungi, but not by mammals in vivo due to the lack of suitable enzymes (Vert), and it can polymerize with propylene glycol to produce a copolymer with a long degradation time. The biopolymer undergoes hydrolytic degradation at an early stage and continues to complete degradation by a surface-to-body process. Manipulation of the copolymer composition is important for the development of ideal controlled release carriers for drugs in vivo (Göpferich et al.).
Table 7-2 Degradation times of biopolymers and their copolymers
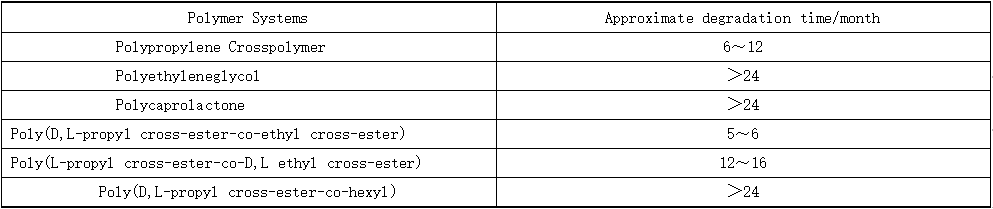
Molecular weight and crystallinity are important factors affecting the degradation of polymers. Studies on PLA (Tsuji, Miyauchi; Zhou et al.; Itävaara et al.; Södergård, Näsman) have shown that the crystalline fraction of PLA is more resistant to degradation than the amorphous fraction. Tuji and MiyaCii found that even if there are amorphous regions between the crystalline regions, they have better hydrolysis resistance than the completely amorphous regions in amorphous PLA. Hydrolysis is the primary stage of microbial and enzymatic degradation, as the hydrolysis of large molecules provides a larger surface area for further effective reactions. Typical polymer crystallization also depends on the composition of the copolymer. The ethyl cross ester in the polypropylene cross ester reduces the crystallinity of the copolymer because the different sizes of the monomer molecules prevent the rearrangement of chains in the compact crystal structure. Gilding and Reed found that while both pure poly(lactic acid) and poly(ethanolide) have a crystallinity of about 35% to 55%, poly(propyleneglycolate-ethyleneglycolate) containing 25% to 65% (molar fraction) of ethyleneglycolate is still amorphous (Figure 7-1). Amorphous copolymers can be used as drug delivery carriers, and simultaneous mass loss in all parts of the carrier can promote good dispersion of the active ingredients of the drug. For sutures or physical implants, where mechanical strength is required, the composition of the ethylene cross ester should be reduced in order to still have good physical properties over time (Gilding, Reed).
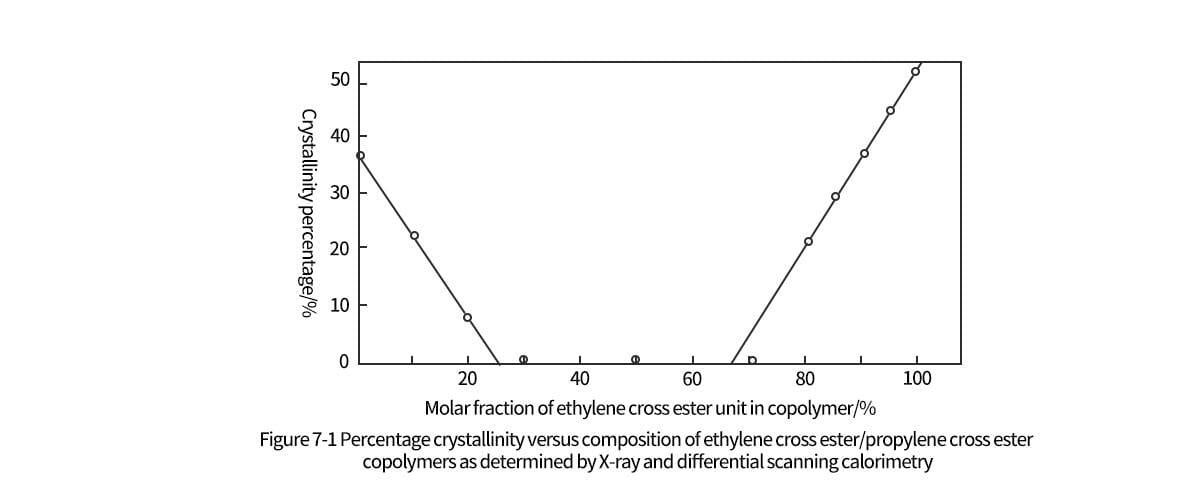
As mentioned earlier, in addition to crystallinity, molecular weight size also plays an important role in the degradation of PLA. In fact, most of the drug/medical grades of PLA are classified according to their characteristic viscosity, which is an indirect method of determining molecular weight. Characteristic viscosity measurements are easier to use for quality control than chromatographic methods, which are more expensive and time-consuming. However, when dealing with sensitive products, characteristic viscosity measurements should be used with caution to ensure the accuracy of the measurement. Researchers have observed a slower degradation rate of high molecular weight polyesters (Saha, Tsuji; Burkersroda et al.). This is due to the fact that high molecular weight molecules have greater entanglement, which means that they resist hydrolysis and thus avoid chain cleavage. The initially surface degraded oligomers tend to form an intermediate medium and are metabolized by living cells and microorganisms (Toki wa, Calabia).
Water absorption and acidity are also important factors affecting the degradation of biopolymers. Usually, water absorption is associated with hydrolytic degradation, in which water molecules react against the polymer during hydrolytic degradation, thus fragmenting it; this is also known as reverse condensation (Södergård et al.). The process of water uptake hydrolysis is an important mechanism to ensure the function of biopolymers in biological systems and their eventual degradation by microorganisms. The degree of water absorption depends on the morphology, molecular weight, purity, sample shape and processing history of the polymer. For example, the crystal structure can reduce the ability of water to penetrate. This can be achieved through copolymerization or quenching processes of the polymer. The water absorption of aliphatic polyesters leads to the breaking of ester bonds, which allows oligomers to be taken up by living cells. Acidity controls the rate of ester cleavage reaction through catalysis (Vert et al.). By comparing polyglycolic acid and poly(ethylene lactate) sutures, Chu found that the fracture strength of the entire suture depended on the pH value, especially at high and low pH values. Under acidic and alkaline conditions, ion exchange can be effectively used to promote the stabilization of chain cleavage.
According to Hutmacher, initially, the hydrolysis process occurs in the first 6 months, during which mass loss occurs while molecular weight remains constant. Excess water is required to penetrate into the higher molecular weight structures to initiate hydrolysis reactions with the ester bonds. After prolonged accumulation of water in the polymer, the ester bond is cleaved and water-soluble monomer oligomers are formed. Lactic acid monomers are formed, which cause hydrophobic degradation over a period of 6 to 9 months. These monomers diffuse into body fluids, resulting in significant mass loss. The broken down monomers and oligomers are further transferred to the liver for metabolism. At this stage, lactic acid in body fluids is subjected to enzymatic degradation, but this is limited to L-lactate, as the body does not produce D-lactase. Therefore, D-lactate takes a long time to undergo hydrolytic degradation and finally breaks down into carbon dioxide and water, which is then excreted from the body.
7.3 Hydrolysis and enzymatic degradation of polylactic acid
Hydrolysis, also known as hydrolytic degradation, is the main degradation mechanism of PLA. It is an autocatalytic process that produces carboxylic acids, i.e., lactic acid contributes to the catalytic hydrolysis process. This was observed in a study in which a thick sample was immersed in a buffer of pH="7.4" at 37°C; subject hydrolysis occurred at a higher rate than surface hydrolysis (Henton et al.). This can be explained by the contact of the surface of the thick PLA sample with the buffer, the easy diffusion of the lactic acid produced by the hydrolysis of the PLA end groups on the surface, and the maintenance of the external pH at pH="7.4." However, the internal cleavage of PLA samples at a higher rate is due to the lower diffusion rate of the resulting acid in the buffer medium; therefore, the lactic acid accumulated from the cleaved PLA end groups induces an autocatalytic reaction.
Hydrolysis of PLA was studied in an aqueous medium such as phosphate buffer solution or water at 37°C to simulate its degradation process in body fluids at appropriate temperatures. To determine the effect of hydrolysis of PLA under harsh and accelerated conditions, the addition of wake-acidic, basic or buffered solutions, and at higher temperatures were also investigated (Tsuji et al.). When crystalline PLLA was hydrolyzed at temperatures below its melting point, it was found that the amorphous zone suffered a significant loss and maintained a crystalline chain, regardless of the hydrolysis medium.
Tsuji studied the hydrolysis of the amorphous form of PLA to determine the effect of L-propyl cross-ester content, stereospecificity, and enantiomeric polymer blends. In this work, four samples were prepared, namely, PDLLA, PLLA, PDLA membranes, and a mixture of PLLA and PDLA samples.
Tsuji found that the copolymers of PDLLA showed significant mass loss compared to the homopolymers either PDLA or PLLA, as shown in Figure 7-2. When PDLA and PLLA are co-mingled, the mass loss due to hydrolysis is not significant. The mass loss of PDLLA copolymers is mainly due to molecular rearrangement, which disrupts the crystalline dense structure associated with the degree of stereospecificity. The PDLA and PLLA copolymer consists entirely of isotropic structures. However, PDLLA consists mainly of isotropic sequences and a small amount of acyclic sequences. As a result, water molecules can easily migrate between the disordered helical conformations of the DL chains and interact efficiently with the sample interior to promote autocatalysis. Although mixing with the stereocomposite homopolymer affects the structural rearrangement of the polymer chains, the mass of the polymer remains unchanged for 24 months. This suggests that pure homopolymers have strong structural properties that prevent hydrolysis from occurring, while water-soluble oligomers formed between mixed regions are thought to be trapped between structures with strong interactions (Tsuji, 2002).
Although there was no significant loss of mass of the PLA films over time, the average molecular weight Mn changed significantly. The poly (D, L-propyleneglyceride) copolymer showed a dramatic change in Mn over 16 months, while the homopolymer and its copolymer decreased gradually. On the one hand, this provides evidence of a weaker copolymer structure, allowing the free migration of water molecules while inducing autocatalytic reactions, which leads to the aggregation of catalytic oligomers formed by hydrolysis. On the other hand, the homopolymer has a strong migration-limiting structure, which allows the hydrolysis reaction to proceed in a mild manner.
PLLA, PDLA and copolymers composed of homopolymers form single crystals when crystallized, while copolymers PDLLA form composite crystals. This suggests that hydrolysis is a chain cleavage process that reduces the molecular weight of the polymer, resulting in oligomers that reduce the formation of microcrystals. The results showed that the Tg, Tc and Tm, H values of the PLLA and PDLA blends were higher than those of the other blends. This is consistent with the molecular weight of Mn, and the molecular weight distribution Mw/Mn remains constant after 24 months of hydrolysis. The results showed that the co-blending of PLLA with PDLA had a significant strong interaction and delayed the hydrolysis reaction. This phenomenon was observed by Tsuji, who found that good steric coordination of poly(lactic acid) D and L mixtures could promote the generation of three-dimensional network structures in amorphous regions. These findings are relevant to the biomedical application of PLA in the control of implant degradation. It is known that D-lactic acid cannot be broken down by enzymes in vivo, and the higher degree of conformational regularity helps the (D,L-propanediol) copolymer to form extremely strong interactions, thus retarding water degradation.
Tsuji et al. further extended the study on the hydrolysis of PLA at high temperatures. Table 7-3 summarizes the thermal properties of hydrolysis and degradation of PLLA under different conditions. It is clear that for samples degraded above 160 °C, PLLA exhibits cold crystallization and the presence of ∆Hcc. Cold crystallization is the result of rearrangement of amorphous regions into crystalline phases (Wellen, Rabello). PLLA formed a wider amorphous zone by chain cleavage reaction during recrystallization under high temperature hydrolytic degradation. This can be demonstrated from the results of Figure 7-3 Tsuji et al. by comparing the molecular weight Mn of PLLA at 120°C and 160°C. When the temperature increases, the molecules in the amorphous region tend to rearrange into a more stable crystalline state, thus undergoing an exothermic process.
Table 7-3 Thermal properties of hydrolytically degraded poly-L-propyl cross-ester
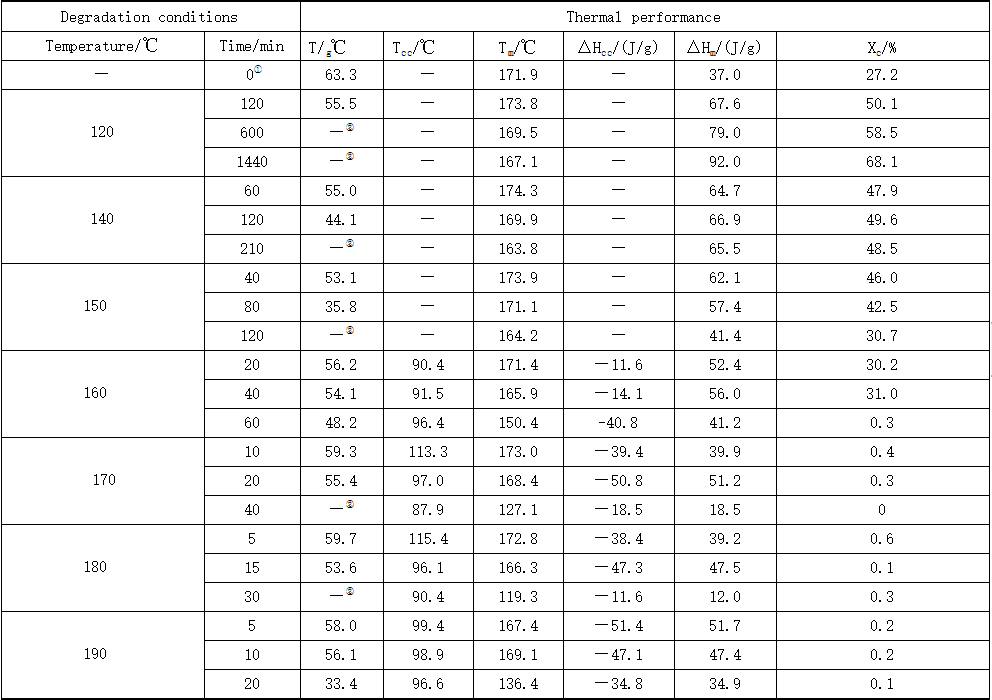
Source: Tsuji et al.
①Before hydrolytic degradation.
②The glass transition is too dispersed to estimate T g.
Nevertheless, water degradation at high temperatures has shown a decrease in PLLA melting temperature (Tm) and percentage crystallinity (Xc) with increasing temperature. Tm and Xc are interrelated:The crystal structure of the polymer requires higher temperatures to induce molecular vibrations (i.e., ∆Hm) in order to liberate it from the lattice. Xc is a measure of polymer crystallinity, including cold crystallization and melt enthalpy, as shown in equation (7-2). According to Miyata and Masuko, the ΔHmc of PLLA at 100% crystallization is 135 J/g. The loss of crystallinity is due to water degradation resulting in a more disordered lattice.
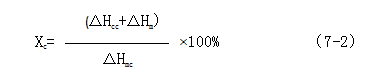
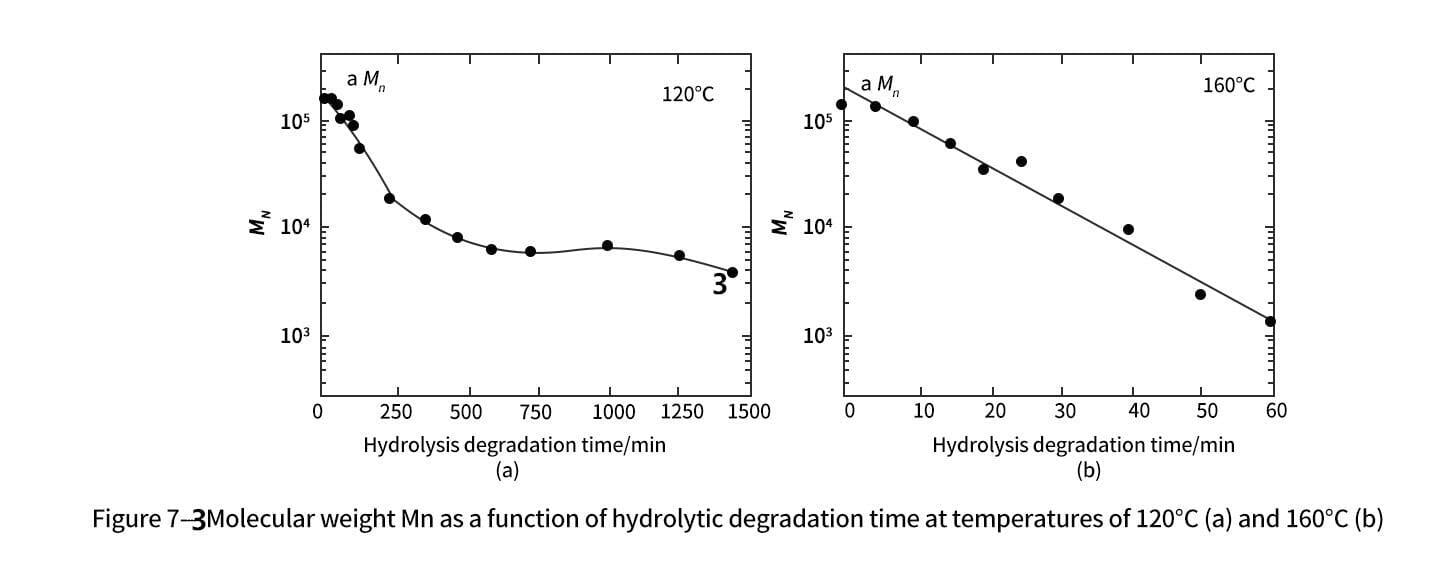
Hydrolytic degradation of PLLA at high temperatures leads to loss of molecular weight and increase in molecular weight distribution. This is similar to the mechanism of bulk erosion found in PLLA. The typical spikes shown in Figure 7-4 were obtained from gel permeation chromatography (GPC) of PLLA after hydrokeratolytic degradation, and the broad peak width indicates a wide molecular weight distribution. The hydrolyzed PLLA in oligomeric form is further reduced to lactic acid as shown in Figure 7-4, where the formation of lactic acid from PLLA hydrolysis gradually increases and the time to reach the total lactic acid yield depends on the temperature. Tsuji et al. showed that lactic acid was successfully obtained at 120°C, 140°C, 160°C and 180°C when degradation lasted for more than 4320 min, 510 min, 180 min and 120 min.
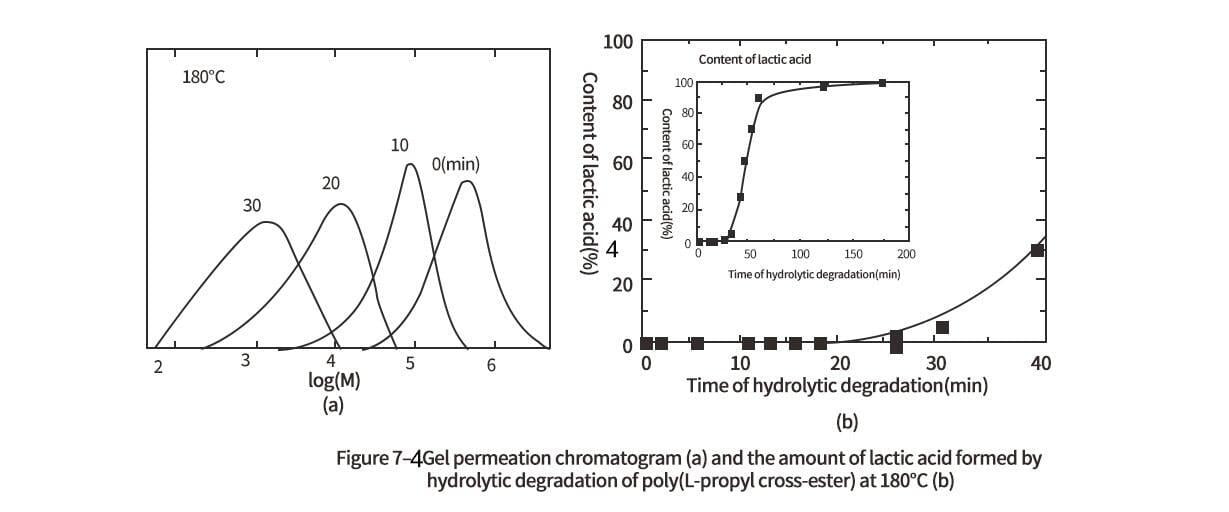
7.4 Environmental degradation of PLA
Understanding the environmental degradation of PLA is important, as more than 100,000 tons of PLA are produced each year, primarily for consumer products and packaging. Therefore, after a short period of use, most PLA will be disposed of in landfills.PLA biodegrades through aerobic and anaerobic pathways and is dependent on oxygen for bacterial and fungal assimilation. There are many ways to measure the biodegradation of biopolymers, such as oxygen consumption, mass loss, biogas generation and CO2 production.
Some of the material properties of PLA including molecular weight, complexity of the building blocks, and crystallinity can affect its degradation. Also, external factors, such as moisture, sunlight, temperature, presence of solvents, and oxygen availability, can significantly affect its biodegradation rate. Massardier-Nageotte et al. studied aerobic and anaerobic biodegradation of commercially available plastics. The results are shown in Table 7-4. The starch-rich MaterBi samples showed the greatest mass loss under both aerobic and anaerobic conditions, while PLA showed a greater mass loss under aerobic conditions and a smaller mass loss under anaerobic conditions. Detailed analysis of MaterBi and PCL samples revealed that PCL had low biodegradability, while the mass loss of MaterBi samples was mainly caused by starch. This is because starch is a natural material that is easily consumed by microorganisms and can be broken down into monomers for consumption without undergoing hydrolysis. In other words, the biodegradation of polymers depends not only on the reactivity of the microorganism to the polymer itself, but also on the chemical degradation of the polymer before it is consumed by living cells that may affect the degradability.
Table 7-4 Mass loss of biopolymers under aerobic and anaerobic conditions
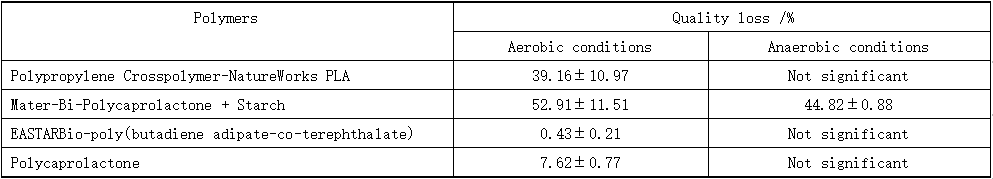
The biodegradation rates of the different biopolymers were further investigated over 7, 14 and 28 incubation days (Massardier Nageotte et al.). The results are presented in Table 7-5.
Table 7-5 Percentage biodegradation of biopolymers under aerobic conditions

PLA appears to have the slowest biodegradation rate among biopolymers. Among the four biopolymers, only MaterBi was able to produce biogas, with 58.2 mL, 113.6 mL and 216.4 mL of biogas per gram of MaterBi sample at 7, 14 and 28 days, respectively. The researchers concluded that PLA biodegrades very slowly compared to other biopolymers and lacks microbial aggregation on the surface of the samples. Typical data show that PLA is more durable and resistant to degradation for a longer period of time than other biopolymers, while maintaining its biodegradable properties. For PLA, it is important to maintain its functionality in a range of applications involving long-term use, such as fabrics and mattresses. These products can be used until they wear out, then biodegraded and eventually converted to harmless residues in the natural environment.
This begs the question, "How long does it take for a PLA product to fully degrade?" Kale et al. investigated the biodegradability of PLA bottles under actual and simulated composting conditions. This study used 500 mL PLA bottles for mineral water sold by Biota, U.S.A. The PLA bottles were manufactured by Nechiwok.The PLA consisted of 96% L-propyl cross ester and blue ketone additive. PLA bottles are composted and buried under controlled conditions in accordance with international standards ASTM D5338 and ISO 14855-1.
When the PLA bottles were buried for 30 days in a compost pile consisting of cow manure, wood chips, and waste feed (i.e., feed left behind by the cows), the bottles had completely decayed by the final stage of testing. Kale et al. reported that the composting temperature reached 65°C due to microbial action and environment, and the high temperature distorted the PLA bottles on the 1st and 2nd days. This is because the temperature is higher than the glass transition temperature (Tg) of PLA (60.6°C). The structure of the bottles then remained solid until days 6 to 9, when a powdery structure appeared on the surface and began to break. By day 15, the bottles had lost their original structure and a large portion had been turned into compost.
Further studies of PLA biodegradation using the cumulative measurement of respiration (CMR) system (according to ASTM D5338 and ISO 14855-1) showed that PLA bottles buried in compost piles require >30 days of biodegradation to reach 80% mineralization. CMR is a system designed to produce a percentage of CO2 from the organic carbon content of a sample. a typical setup of a CMR system is shown in Figure 7-5. It consists of a set of bioreactors with controlled air supply. A 10 mol/L sodium hydroxide (NaOH) solution is passed under pressurized air at 2 psi to measure the CO2 in the air. Deionized water is mainly used to humidify the air When measured using a relative humidity (RH) meter, the humidified air is further mixed with dry air to achieve 50% humidity. Bioreactors containing compost and vermiculite provided a high water holding capacity of 300% compared to only 28% to 45% in sandy to clayey soils (Grima et al.). Samples were placed in compost while cellulose was used as a comparison material, and during the test, microorganisms in the bioreactor consumed the samples and released CO2. CO2 is trapped in a 0.25 mol/L solution of NaOH. A small known amount of the reacting NaOH solution is transferred out and an acid titration (e.g. to HC1 solution) is performed for a certain time to determine the amount of CO2 produced.
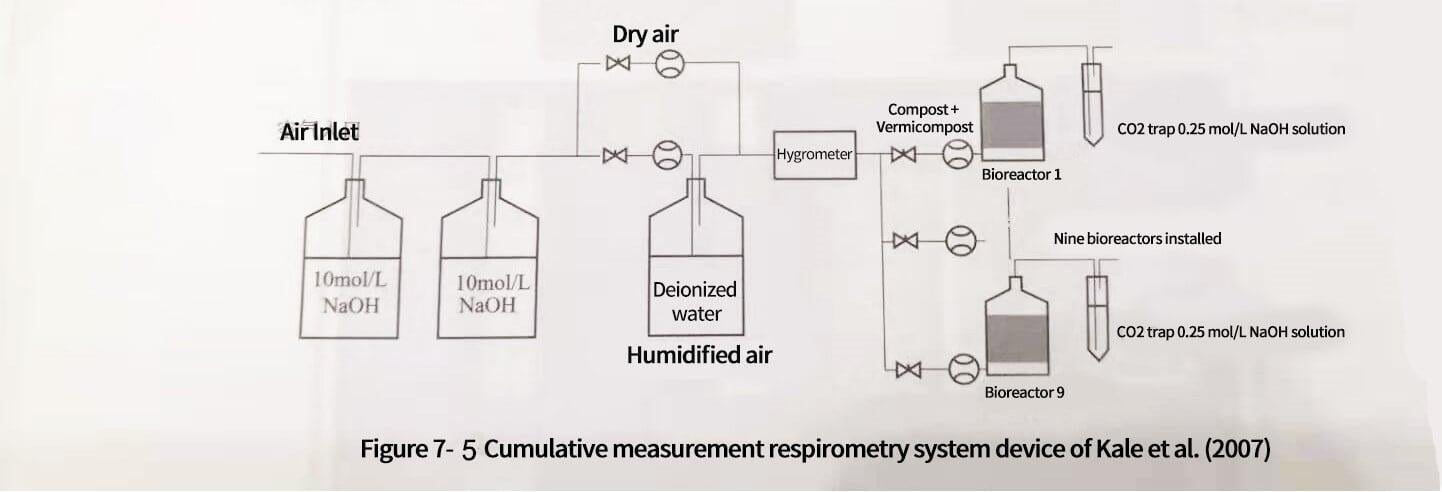
The titration reaction scheme is described in ASTM D5338 as follows, using the strong acid HC1
During the absorption of CO2 from the biodegradation of the sample.

Titration reaction.
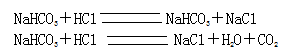
The titration is performed using an indicator such as phenolphthalein. the CO2 concentration can be calculated according to the following equation.
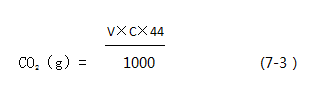
where V is the volume of HC1 consumed in the reaction (R-4). The mineralization rate is used to calculate the amount of CO2 produced in positive controls such as fibers and in blank PLA samples according to the equation.
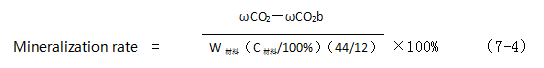
where ωCO2 is the total amount of CO2 in grams generated by the sample and the positive control, and ωCO2b is the total amount of CO2 in grams generated in the blank group; W material = sample mass; C material = organic carbon content of the sample.
Kale et al. also reported the difference in biodegradation rates of PLA and general biopolymers in actual soil burial and simulated compost, as shown in CMR. The high biodegradation rate of the simulated compost was mainly due to the small sample size used in the experiment, which enhanced hydrolysis and provided a larger surface for microbial reactions. Under actual composting conditions, the biodegradation rate tends to be slower due to humidity, composting materials, microbial species and the larger size of the treated products. Therefore, Kale et al. (2007) concluded that true composting trials must be conducted to ensure that biopolymer products can be successfully biodegraded and decomposed in commercial composting facilities and landfills.
The types of microorganisms involved in biodegradation were studied earlier by Torres et al., who used various microbial strains. The objective was to screen for microorganisms involved in the biodegradation of PLA and lactic acid-containing polymers. Initially, the researchers used D,L-lactic acid (D,L-LA) and its oligomers to study the degree of response of filamentous fungi over a 7-day period. Torres et al. (1996) performed two separate analyses of D,L-LA and oligomers at a concentration of 10 g/L and sterilized them to avoid biological contamination that could lead to erroneous results. The results showed that all strains were able to actively consume lactic acid and oligomers. Of the strains analyzed, only three were able to fully utilize D,L-LA and D,L-LA oligomers as the sole source of carbon and energy (two strains of Fusarium cepacia and one strain of Penicillium roqueforti). Other strains can only partially absorb DL-lactate and oligomers. This suggests that lactic acid is only a source of assimilation for some strains. The biomass of Fusarium cepacia and Penicillium roqueforti remains high and the material produced by the strains assimilation has been a good source of plant nutrients.
7.5 Thermal degradation of PLA
Polymeric materials are typically used above room temperature. Existing general-purpose polymers such as polyethylene, polypropylene, polystyrene and polycarbonate are often used to make cups and containers for hot food and hot drinks, and even for plumbing hot water. For this reason, marketable biodegradable PLAs should also have similar thermal stability characteristics in order for PLAs to replace existing commercial polymers and be more widely processed and used.
The thermal degradation of PLA was originally studied by McNeil and Leiper using thermogravimetry. The highest degradation rate of PLA has been reported at 365°C under nitrogen gas flow, with accelerated decomposition in excess air due to oxidation by free oxygen. This was also found in a recent study by Zhan et al., which also compared the fire resistance of PLA after the addition of flame retardants SPDPM is an intumescent flame retardant. PLA had a simple single-stage degradation, with an initial mass loss of 5% occurring at 325°C and no residual at the final heating to 500°C.
Carrasco et al. investigated the thermal decomposition and stability of PLA as they relate to processing methods. Table 7-6 provides a summary of this work:PLA-V is newly supplied by the manufacturer with the highest degradation temperature (Tn,n=5, 50, 95, indicating the percentage mass loss and p is the highest decomposition rate at the corresponding temperature). As PLA is extruded and injected, thermal degradation causes a slight decrease in molecular weight. This is due to repeated heating and cooling, which resulted in a slight decrease in molecular weight, and the presence of moisture caused a hydrolysis reaction. Wang et al. found that when PLA samples were extruded/injected, they had more chromogenic groups, i.e., the presence of double-terminal bond CCs, and conjugates with the carbonyl group (CO), which triggered chain breaks and led to yellow coloration. Table 7-6 further shows that the annealed PLA samples have lower degradation temperatures. This may be due to the reaction of intramolecular functional groups caused by prolonged exposure to high temperatures, although the annealing process resulted in PLA crystallization and a slight increase in the glass transition temperature. When used for hot food and hot beverage containers, a higher glass transition temperature is advantageous because This avoids collapse of the container due to polymer softening.
Table 7-6 Thermal degradation properties of polylactic acid (PLA) in the corresponding processing methods analyzed by TGA
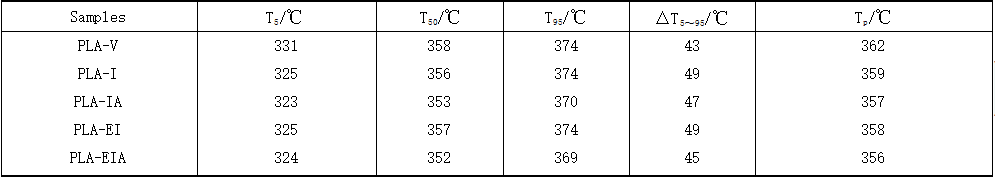
Note: PLA-V-unprocessed material; PLA-I-injection; PLA-EI-extrusion and injection; PLA-IA-injection and annealing; PLA-EIA-extrusion, injection and annealing.
The thermal stability is linearly related to the average molecular weight. In fact, the initial decomposition temperature T5 increased by 2.6°C for every 10000 increase in number-average molecular weight (Mn) (Figure 7-6), while T5 increased by 1.4°C for every 10000 increase in heavy-average molecular weight (Mw). Secondly, thermal and shear processing of PLA during extrusion and injection leads to lower molecular weight, which is the main reason for the weakened thermal resistance of PLA. This was further confirmed by comparing the polydispersity index measured for PLA-EI (after extrusion and injection) and PLA-I (after injection) (Figure 7-7): the initial decomposition temperature of PLA-EI was lower than that of PLA-I. In short, careful selection of reworking and processing methods for PLA is essential to maintain the inherent properties of PLA.
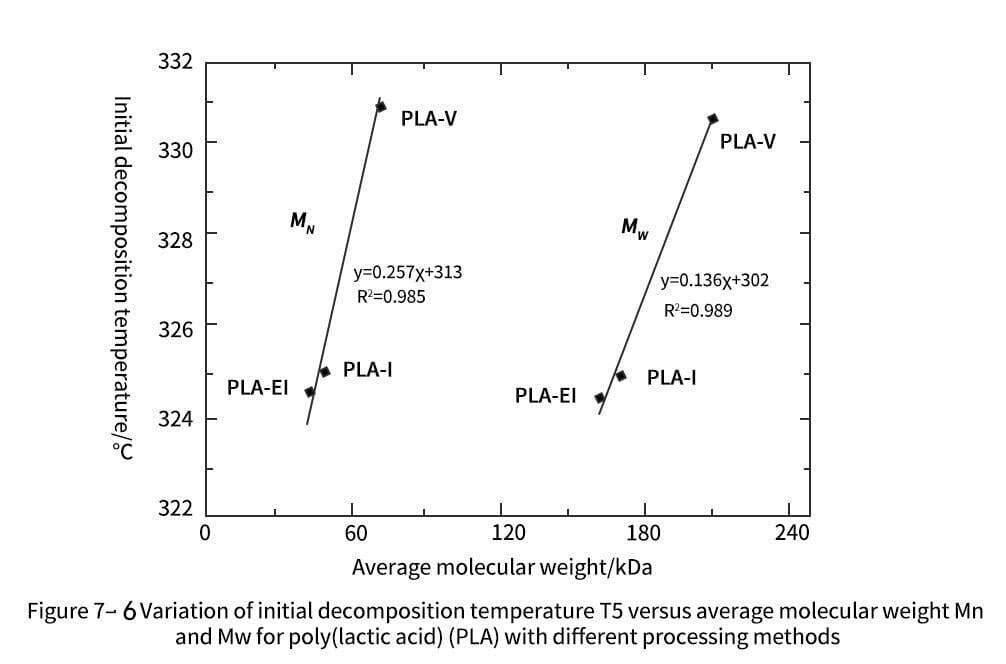
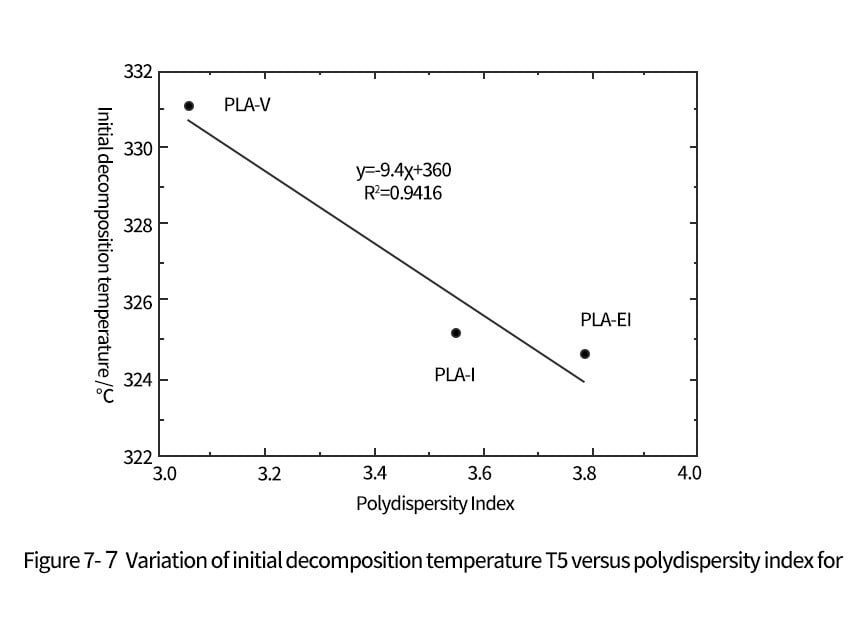
For some outdoor applications, photodegradation can have a significant impact on the life of PLA products. In general, when polymers are exposed to outdoor environments, they are subject to weathering effects. Ultraviolet (UV) light and moisture are the main degradation agents that lead to changes in the chemical structure of the polymer, which further affects its mechanical properties. When PLA is subjected to accelerated aging under UV light, its chemical structure undergoes essential changes, including chain breaking, cross-linking and intermolecular reactions to form new functional groups.
The elution map for GPC analysis reported by Belbachir et al. showed that the elution of pristine PLA increased under UV-accelerated irradiation.
In addition, the elution profile of irradiated PLA also shows a wider curve compared to the original PLA. This indicates that the molecular weight distribution is broadened due to the chain-breaking effect. At higher UV doses, the molecular weight starts to decrease while the molecular weight distribution increases further. Free radicals are generated during photodegradation, invading the backbone and forming lower molecular weight products. Ikada proposed that PLA follows the Norrish II mechanism, which involves extensive chain cleavage and the formation of CC and OH, as shown in Figure 7-8.

When UV reaches the backbone of PLA, electronegative oxygen atoms are activated to form free radicals, which is known as photophysical excitation. The attack reaction also includes free oxygen coming from the air, which eventually cracks the PLA molecular chains. Long-term exposure of PLA to UV irradiation leads to loss of its mechanical properties. The almost linear relationship between UV dose and changes in elastic modulus and yield stress, while the stress-strain at fracture showed a rapid decrease at higher UV doses.This can be explained by the fact that higher UV triggers chain cracking and generates excessive local cavitation, which promotes the generation of local weak points. When external loads are applied to the irradiated sample, these weak points transmit and merge, damaging the whole structure.
Photosensitizers can be added to enhance the photodegradation of PLA with the aim of increasing the degradation rate during accelerated PLA waste treatment. Tsuji et al. investigated the effect of N,N,N′,N′-tetramethyl-1,4-phenylenediamine (TMPD) on amorphous and crystalline films of PLA. When TMPD is exposed to UV irradiation, it is activated and releases free radicals that attack the backbone; the mechanism is similar to the Norrish II reaction.The photodegradation of PLA was promoted by TMPD, independent of the crystallinity of PLA. This suggests that the formation of free radicals is related to the free reaction of the backbone, whereas hydrolytic degradation requires the contact of water molecules with the amorphous structure for chain cleavage to occur (Tsuji et al.).
However, electron irradiation has also been used to improve the physical and mechanical properties of PLA. Improving the structural properties of PLA is very important, especially for thermoformed PLA products that are used at high temperatures. This is because pure PLA is likely to be softened at temperatures above 60°C. The electron irradiation of PLA requires triene isocyanurate (TAIC) (Figure 7-9) as a cross-linking agent to enhance its properties.

Without it, electron irradiation tends to degrade PLA, deteriorating both the processing properties and the functional quality of PLA (Malinowski et al., Kanazawa). Figure 7-10(a) shows that the melt flow rate (MFI) of pure PLA decreases with increasing electron irradiation dose. mFI is a measure of viscosity; higher MFI means lower viscosity. High doses of electron irradiation cause chain breakage, which leads to a decrease in molecular weight and viscosity of PLA. Different phenomena can be observed after the addition of TAIC. With the addition of 1% TAIC, the MFI is improved but still lower than that of pure PLA. this indicates that chain cracking and cross-linking occur simultaneously. According to Malinowski et al., the amount of TAIC should be less than 1% because excessive TAIC causes thermal curing of PLA, which prevents the material from extruding through the extruder head. However, electron crosslinking can improve the glass transition temperature (Tg) of PLA (Figure 7-11). The maximum value of Tg can be achieved at a dose of 60 kGy with the addition of 3% TAIC. This cross-linking method can be used to make film products suitable for PLA thermoforming when high temperature applications are required.
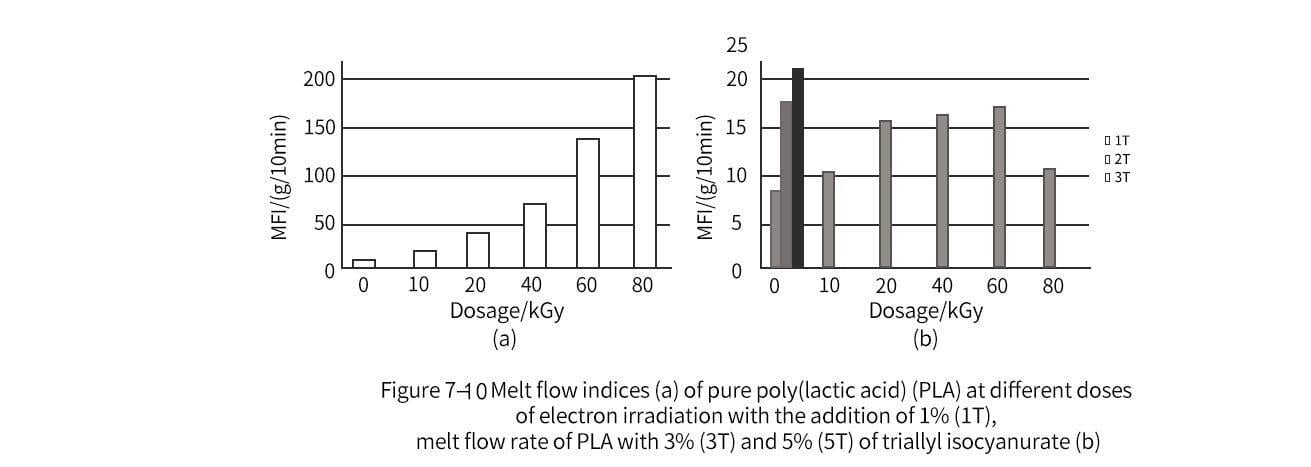

7.6 Flame retardancy of PLA
When electronic and electrical equipment is subjected to current and voltage for extended periods of time, some misuse or malfunction can lead to accidental ignition, such as a short circuit event. The flammability of PLA is important for fire risk reduction as the application of PLA has been extended to enclosures for electrical and electronic equipment. In addition, understanding the combustion behavior of PLA can help design flame-retardant packaging that meets fire safety requirements and maintains its biodegradable properties.
The flammability of plastic materials should be evaluated according to UL-94 and the limiting oxygen index (LOI). UL-94 is the best known standard, published by Underwriters Laboratories. According to this standard, plastics are classified according to their combustion characteristics and further divided into 12 flame categories. Basically, consumer electronics used to manufacture enclosures, structural parts and insulators are classified into six classes 5VA, 5VB, V-0, V-1, V-2 and HB. Ratings HF-1, HF-2 and HBF are used for low density foam materials in loudspeaker grilles and anechoic materials, and the last three ratings VTM-0, VTM-1 and VTM-2 are used for very thin films.
LOI is used to determine the minimum percentage concentration of oxygen required to sustain polymer combustion. The flow of nitrogen is controlled during the combustion of the specimen until a critical level of oxygen is reached. LOI is measured according to:BS EN ISO 4589-2 Plastics. Determination of oxygen index burning properties, ambient temperature test and measurement of the minimum oxygen concentration to support candlelight burning of plastics ASTM D2863-10 standard test method (oxygen index). According to the experimental reports of Réti et al. and Zhan et al., because of the flammability of PLA, none of the grades of pure PLA meets the standard UL-94, it has a large amount of burning droplets, which are dangerous when dripping during combustion and cause burns when it comes in contact with the skin. In addition, flame-carrying drops can allow a fire to spread further from its initial source to another area, providing a source of ignition and fuel to ignite a new fire. To meet the requirements of fire safety, the development of PLA packaging flame retardants is essential.
It has been suggested that PLA should use intumescent flame retardant technology. Intumescent technology is a passive fire protection technology. Flammable polymer materials produce a light carbon that acts as a poor conductor of heat retarding heat transfer. As shown in Figure 7-12, the carbon barrier uses surface insulation foam to separate combustible materials from sources of ignition, heat, and oxygen. The carbonized layer acts as a physical barrier, effectively reducing heat and mass transfer between the gas and the condensed phase.
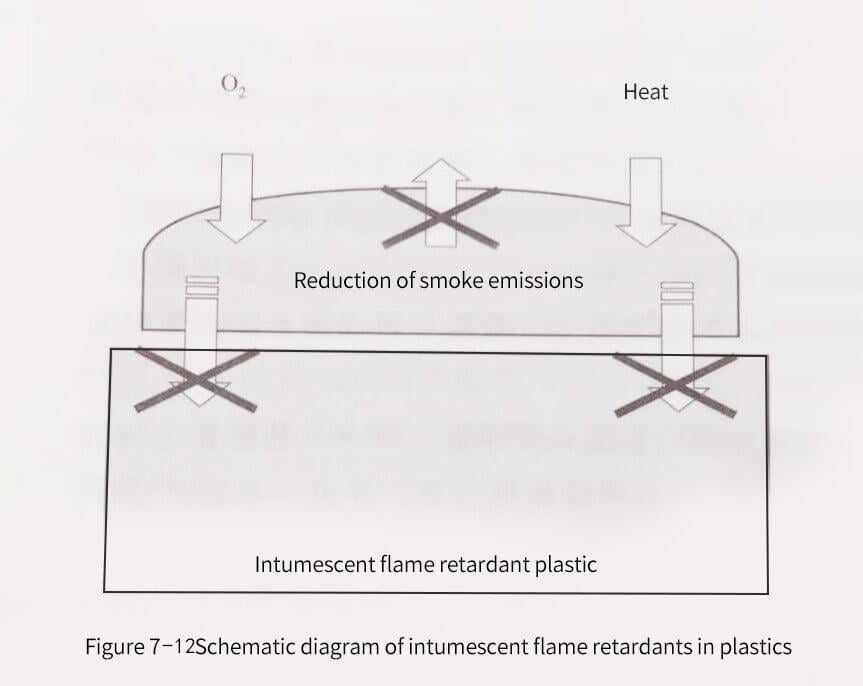
Typical swelling technology flame retardant systems usually consist of acids, ammonium salts and phosphates. Réti et al. evaluated the efficiency of an intumescent flame retardant system using ammonium polyphosphate (APP) and pentaerythritol (PER) in PLA. In this system, APP is both an acid source and a blowing agent, while PER is a charring agent. APP decomposes at high temperatures to produce phosphoric acid derivatives, which act as catalysts to accelerate the decomposition of PER and form coal coke. During APP decomposition, the low boiling point acid derivatives act as foaming agents to produce non-combustible gases that expand the coke layer. However, APP and PER are petrochemicals and are not biodegradable. Mixing APP and PER with PLA will reduce the "green" qualification of PLA. Therefore, Réti et al. used starch and lignin as carbonization agents instead of PER. Wang et al. also reported an improvement in LOI with increasing starch content, suggesting that higher concentrations of oxygen are required to maintain combustion (Figure 7-13). This finding favors the blending of starch with PLA because it is flame retardant without affecting its biodegradability.
Zhan et al. observed that the formation of coal coke was very similar when spirocyclic pentaerythritol diphosphoric acid diphosphoryl melamine flame retardant (SPDRM FR) was added to PLA. This observation suggests that starch has a swelling effect comparable to that of synthetic flame retardants.
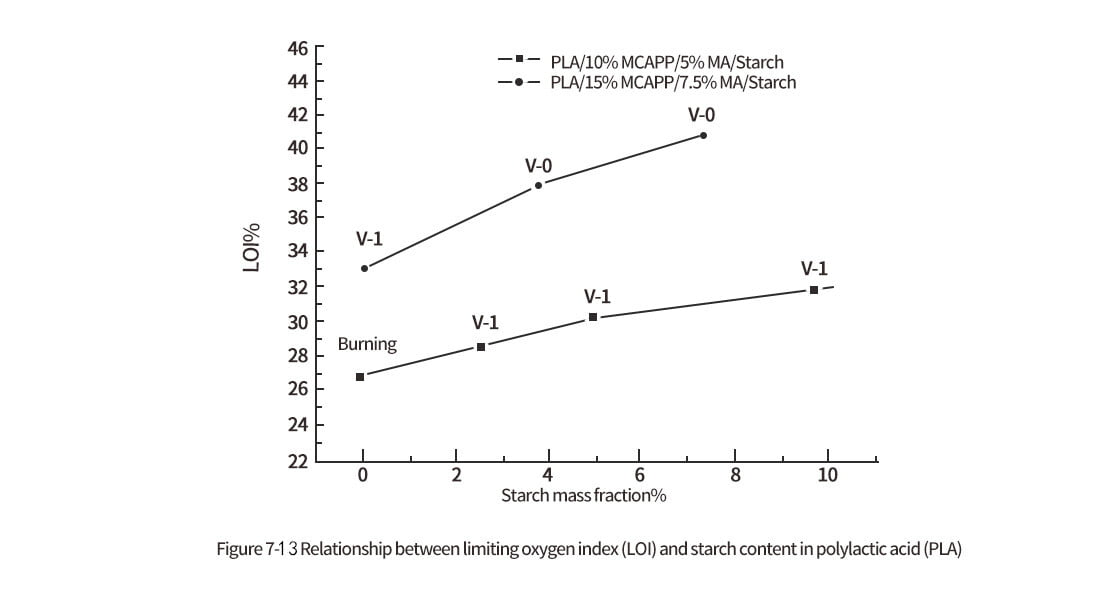
Li et al. used organically modified montmorillonite (OMMT) in combination with APP as an intumescent flame retardant for PLA. According to these researchers and Si et al., the purpose of OMMT is twofold: (i) the formation of carbonaceous silicate coal coke on the polymer surface during combustion protects the polymer matrix and slows the mass loss rate; and (ii) MMT provides an anti-drip effect to the burning polymer.
Compared to branched or thermoset polymers, linear polymers such as PLA, polyethylene terephthalate and PBS have a lower melt viscosity, so their anti-drip effect plays a very important role in these linear polymers. These polymers are very unfavorable when it comes to combustion: they exacerbate combustion by producing severe melt dripping. Li et al. successfully overcame the dripping of PLA with N,N-dimethyl dehydrogenated tallow quaternary ammonium salt-modified MMT and APP flame retardant systems co-blended with PLA. Adding MMT and combining it with an intumescent flame retardant is critical because neither of these additives (MMT and intumescent flame retardant) can control the dripping of PLA in fire alone. However, APP is an effective flame retardant with the highest LOI even without the addition of OMMT. In conclusion, it is important to choose the right flame retardant to inhibit the flammability of PLA, especially when fire safety is required.
7.7 Conclusion
Polylactic acid is a biodegradable polymer of agricultural origin. These characteristics need to be further enhanced for the diverse applications of PLA, whose biodegradability and biocompatibility mean that it has a wide range of biomedical applications. The rate of PLA biodegradation in vitro and in vivo can be influenced by controlling the crystallinity and the copolymerization reaction of isomers or other monomers. Typically, the degradation of PLA begins with a hydrolysis process followed by enzymatic or microbial action. This degradation eventually leads to the breakdown of PLA and its conversion to harmless substances.
The low softening point of PLA limits its use at high temperatures. However, cross-linking, copolymerization and recrystallization all help to improve its thermal properties. Long-term exposure to heat radiation and UV radiation can cause severe degradation of PLA. In terms of fire protection, PLA is a combustible material and the selection of flame retardants can improve its fire performance in electrical and electronic applications. In summary, understanding the degradation and stability of PLA is an important preparatory step in controlling its performance while maintaining its "green" properties.
8. Additives and processing aids for polylactic acid
8.1 Limitations of PLA in processing and application
Currently, polylactic acid (PLA) is one of the most widely used biodegradable polymers and can replace non-biodegradable petroleum-based polymers in many applications such as food packaging and biomedical devices. In addition, PLA has many advantages such as being environmentally friendly and biocompatible. PLA is biodegradable, recyclable, and can be obtained from renewable resources such as wheat, rice, and corn (Farah et al). In addition, the biocompatibility of PLA with human tissues has received widespread attention in the biomedical field because PLA is non-toxic and non-carcinogenic to human tissues. After implantation, it was also found that the implanted PLA devices produced only non-toxic degradable products during the degradation process without interfering with the tissue healing process. However, PLA still has some drawbacks that limit its application in food packaging and biomedical applications. The poor mechanical properties of PLA, such as low tensile strength and modulus, brittleness and low elongation at break, are the main reasons that limit the widespread use of PLA in industry (Farah etal.). PLA is a brittle polymer with a low elongation capacity under strain. The poor toughness of PLA greatly limits its application in the food packaging and biomedical industries. For example, the use of PLA as a material for assembling screws and fracture fixation plates is limited in orthopedic surgery. This is because PLA biomedical components or devices, such as screws or fracture fixation plates, require a high plastic deformation capacity under high stress level conditions (Daniels et al.). The low stiffness of PLA implant devices can cause excessive bone movement and impede the healing process.
In addition, the slow degradation rate of PLA is one of the important reasons limiting its application. PLA is a synthetic polymer with hydrolytically unstable ester functional groups in the main chain of the polymer chain. Biodegradation of PLA initially occurs by hydrolysis of the main chain ester functional group to form soluble oligomers with a dramatic decrease in molecular weight (Farah et al.). Finally, soluble oligomers are metabolized by the body. the rate of PLA biodegradation depends on the crystallinity of PLA, molecular weight, molecular weight distribution, the rate of water penetration through the polymer matrix, and the composition of the stereoisomers (Rasal et al). Crystalline regions of PLA polymers were found to be more resistant to degradation compared to amorphous regions (Tokiwa, Calabia). The biodegradation behavior of PLA is its most important feature and has attracted great interest and interest in applications in the biomedical industry. This is attributed to the slow degradation time of PLA, which can extend the life of the implanted device or devices in the body, making it possible in some cases up to several years (Bergsma et al; Rasal et al.). Several reports mention that PLA-based implanted devices require a second surgery to remove the device three years after the implantation procedure (Incardona et al.; Bergsma et al.). the slow degradation rate of PLA and the years required for implant degradation hinder the use of PLA-based implanted devices in biomedical applications.
The poor processing of PLA limits its application in certain industries. PLA can be processed in different ways depending on the application, including extrusion, compounding, blow molding, and injection molding (Farah et al). However, processing at high temperatures (≥200°C) or longer processing times can severely reduce the thermal stability of PLA by reducing the molecular weight and changing the physical properties of PLA through oxidative backbone breakage (Ng et al.), hydrolysis, reforming of propyl cross-ester, and inter- or intra-molecular reactions of ester exchange. For example, the processing temperature required for PLA homopolymers is about 185-190°C, which is too high compared to the melting temperature of 175°C (Farah et al.).Auras et al. found that PLA degrades severely when processed at a processing temperature 10°C above the melting temperature. At these processing temperatures, both shearing and cleavage of chains in the PLA matrix lead to a decrease in PLA molecular weight, which results in thermal degradation (Carrasco et al.; Farah et al.). According to Carrasco et al., processing leads to a decrease in the molecular weight of PLA, which leads to a decrease in mechanical properties. In addition, they also found by performing X-ray diffraction (XRD) and DSC analysis that mechanical processing leads to the loss of crystalline structures in the PLA matrix. This is due to the rapid cooling time after extrusion, blow molding, injection molding, etc., which results in the polymer chains not being able to rearrange into a crystalline structure and only remaining as disordered structures (Migliaresi et al.). Their observations also conclude that processing of PLA severely reduces the crystallinity of the PLA matrix.
In addition, the hydrophobic behavior of PLA may lead to a low affinity between cells and PLA-based materials and cause an inflammatory response in living organisms upon direct contact with biological fluids (Rasal et al.). This is a major problem for the use of PLA in biomedical implantable devices and plates. This is due to the fact that its hydrophobicity causes the required biological activities to not be carried out on PLA. In addition, the lack of functional groups in PLA also leads to bulk and surface modification of the material. (Kakinoki, Yamaoka).
8.2 Toughening of PLA
In order to improve the mechanical properties, such as stiffness, of PLA nanocomposites, various studies have been conducted by adding various types of reinforcing fillers, including montmorillonite (MMT), talc, and carbon nanotubes (CNT), to PLA nanocomposites (Balakrishnan et al.; Lee et al.). It was observed that the addition of MMT was effective in improving the stiffness (Young's modulus and flexural modulus) of PLA nanocomposites. In a study conducted by Balakrishnan et al., it was found that the addition of 4 parts by mass of MMT particles to PLA increased the Young's modulus and flexural modulus by 10% and 18%, respectively. According to Jiang et al., the addition of 7.5% (all mass fractions in this chapter) of MMT particles to PLA nanocomposites resulted in a rapid increase of Young's modulus by 43%. The improved stiffness (Young's modulus and flexural modulus) of PLA nanocomposites is mainly attributed to the effective intercalation/exfoliation of the MMT stacked layers in the PLA matrix. This is because effective intercalation/exfoliation of MMT particles in PLA substrates leads to an increase in the interface area for their interaction with PLA substrates. Therefore, improving the interaction between MMT particles and PLA substrates leads to an increase in PLA stiffness. The larger interfacial interaction region can further effectively transfer the applied stress from the PLA matrix to the MMT particles, thus improving the stiffness of the PLA nanocomposites. In addition, the intercalated MMT particles may also further restrict the motion of PLA molecular chains. The intercalation effect of MMT particles in PLA matrix can be evaluated by using XRD analysis.
Balakrishnan et al. reported that the increase in the number of MMT particles in the PLA matrix reduced the tensile and flexural strength of PLA composites by 10% and 25%, respectively. This also indicates that too many MMT particles tend to agglomerate and weaken the interfacial bonding between the MMT particles and the PLA matrix. This further leads to the agglomerated MMT particles acting as stress concentration points in the PLA matrix when external stresses are applied and failing to transfer the stresses uniformly throughout the PLA matrix. Moreover, due to the brittle behavior of PLA nanocomposites, the material fails early in the strain process. In addition, the dispersion and orientation of the MMT particles in the PLA matrix also play an important role in the tensile and bending strengths due to the different stress directions applied when doing tensile and bending tests (Balakrishnan et al.). By referring to the results of Balakrishnan et al., it can be seen that increasing the amount of MMT to 4 parts (parts by mass) decreases the impact strength of PLA nanocomposites by 13%, which has a significant negative impact on the impact resistance. However, the addition of 2 and 4 parts of MMT particles increased the impact strength of PLA/LLDPE nanocomposites by 53% and 21%, respectively (Balakrishnan et al.). The addition of 10% (mass fraction) of LLDPE to MMT/PLA nanocomposites improved the impact strength compared to PLA nanocomposites. From this observation, the presence of LLDPE provides better distribution and orientation of the MMT particles in the PLA matrix, thereby increasing the energy absorbed by the polymer matrix during rapid loading.
Harris and Lee found that the addition of 2% (mass fraction) of talc significantly increased the flexural strength and flexural modulus of PLA by 25%. This was due to the fact that the added talc particles acted as nucleating agents to induce PLA crystallization, which further improved the toughness of PLA. In addition, due to the structure of the talc particles and their orientation in the PLA matrix, the presence of talc can also effectively transfer the applied stress from the talc particles to the PLA matrix, thus providing a reinforcing effect on the stiffness and toughness of the PLA matrix. Yu et al. investigated the effect of increasing the talc content on the mechanical properties (flexural strength and modulus) of PLA, as shown in Figure 8-1. They also obtained results similar to those of Harris and Lee, i.e., the addition of talc significantly increased the flexural strength and flexural modulus of pure PLA. In addition, Yu et al. found that the flexural strength and flexural modulus of PLA increased rapidly when the talc content was increased from 0% to 2.0%. The addition of talc filler to improve the flexural strength and flexural modulus of PLA is mainly to use the natural high rigidity talc filler instead of PLA matrix so that it can effectively limit the ductility and flow of PLA matrix when subjected to external load. However, when the talc content was further increased from 2% to 24.3%, only a small increase in flexural strength and modulus was found for the PLA composites. When the talc content was further increased above 2%, the enhancement of flexural strength and modulus of PLA composites by talc was less significant.
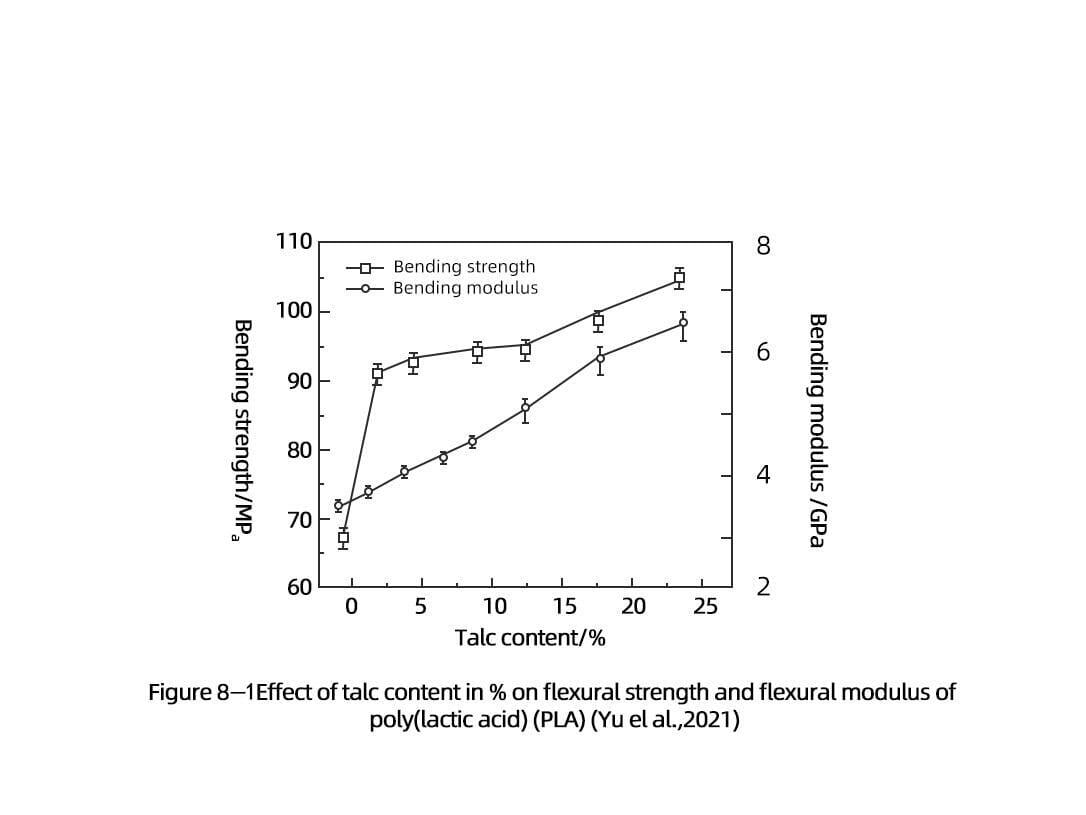
Ouchiar et al. compared and investigated the effect of increasing talc and kaolin content on the properties of PLA composites. The addition of 5% (mass fraction) of talc was found to slightly increase the Young's modulus of pure PLA from 2.4 GPa to 2.6 GPa. In addition, the Young's modulus of the PLA compound material with 5% kaolin added has a similar increment to that of the PLA compound material with 5% talc added. According to Ouchiar et al., further increasing the content of talc and kaolin from 5% to 30% can gradually increase the Young's modulus of PLA composites, as shown in Figure 8-2. However, the Young's modulus increment of PLA composites with talc addition was significantly higher than that of PLA composites with kaolin addition. This is due to the fact that PLA composites with talc added show earlier onset of crystallization compared to pure PLA and PLA composites with kaolin added, thus improving the stiffness of PLA composites, which also confirms the nucleation effect of talc.
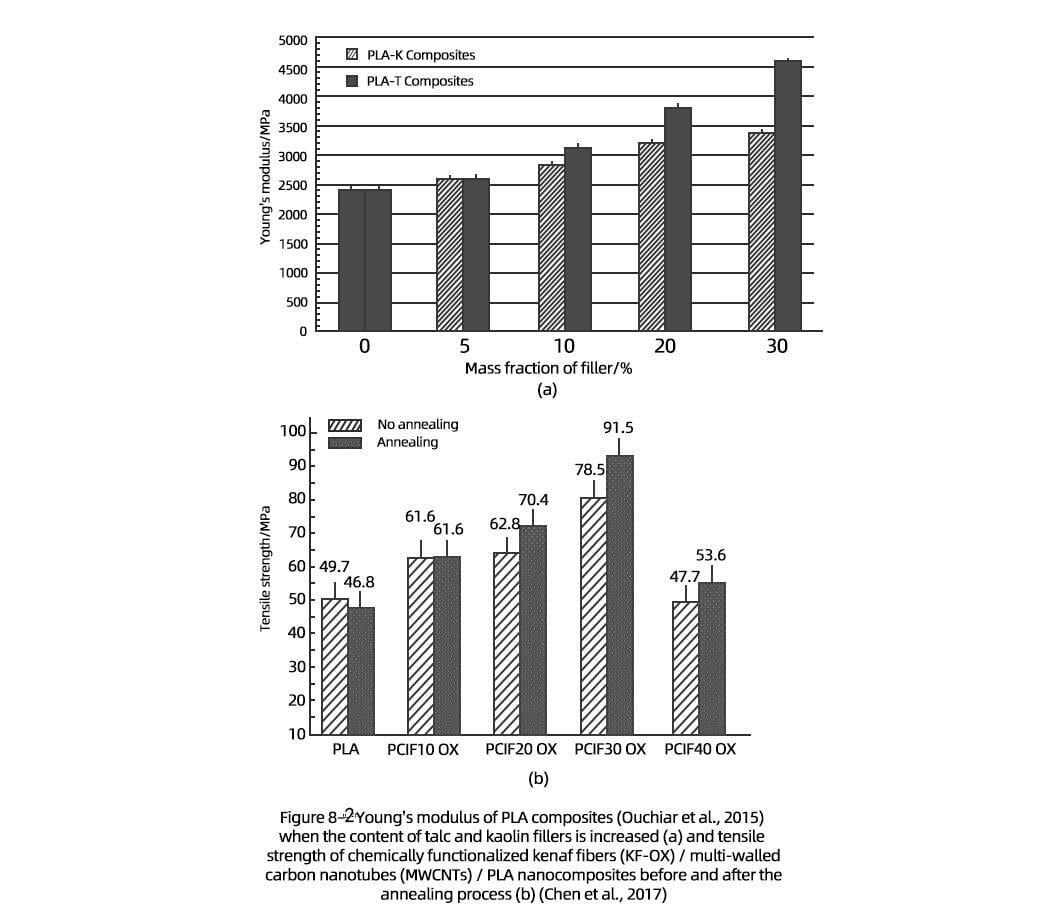
According to Zhou et al., the use of CNTs containing carboxyl groups (CNTs-COOH) (mass fraction of 0.5% or more) can rapidly improve the tensile strength and cantilever beam impact strength of PLA nanocomposites. This also indicates that the addition of small amounts of CNT can improve the tensile strength and impact strength of PLA. This may be due to the high stiffness of CNT with high aspect ratio and surface area, which can be effectively interlocked in the PLA matrix by CNT to further strengthen the PLA matrix. The interlocking effect of CNT particles can effectively transfer the applied stress from the CNT particles to the PLA matrix, thus achieving the reinforcement of PLA nanocomposites. In addition, the strong chemical bond between CNT-COOH particles and PLA matrix hinders the movement of PLA macromolecular chains, thus enhancing the PLA matrix. However, Zhou et al. also reported that increasing the content of CNT-COOH from 0.5% (mass fraction) to 2.0% significantly reduced the tensile strength and cantilever beam impact strength from (42.8 Shi 0.3) MPa and (27.7 Shi 0.5) kJ/m2 to (39.6 Shi 0.2) MPa and (8.8 Shi 0.3) kJ/m2 , respectively. Similar results were found in a study conducted by Wang et al., which also found that the addition of higher levels of CNT (mass fraction > 3%) gradually reduced the tensile strength of PLA nanocomposites. The small decrease in tensile and impact strength may be attributed to van der Waals force interactions between CNT particles, which tend to aggregate into larger CNT agglomerates when the CNT content in the PLA matrix is high (Zhou et al.; Wang et al.). Therefore, the CNT agglomerates in the PLA matrix reduce the adhesion between the CNT and the matrix, and also act as stress concentration points, weakening the transfer of the applied load throughout the PLA matrix.
In a study conducted by Chen et al., they made KF-OX by coupling 3-(methacryloyloxy) propyltrimethoxysilane (OX-silane) to overcome the poor compatibility between hydrophilic kenaf fibers and hydrophobic PLA substrates. Chen et al. added KF-OX and MWCNT to PLA matrix in order to investigate the effect of increasing KF-OX content on the properties of PLA nanocomposites. The tensile strength of MWCNT/PLA nanocomposites was gradually increased by gradually increasing the content of KF-OX up to 30% (mass fraction), both before and after decomposition. The enhancement of the tensile strength of PLA nanocomposites is mainly attributed to the chemical reaction between KF-OX fibers and the PLA matrix. It was also observed that the annealing process recrystallized the PLA nanocomposites, thus improving the tensile strength of the KF-OX/MWCNT/PLA nanocomposites. The addition of higher content of KF-OX (30%) can rapidly increase the tensile strength of PLA nanocomposites to 91.5 MPa (84% higher than pure PLA) when annealing process is performed. This is due to the good compatibility of KF-OC with PLA matrix and the formation of crystalline structure at the interface between PLA matrix and KF-OX fibers, which significantly improves the mechanical properties (tensile strength) of PLA nanocomposites. This is also attributed to the fact that the transverse crystal structure in the PLA matrix increases the adhesion of the interface between the KF-OX fibers and the PLA matrix, thus providing resistance to the applied external loads (Quan et al.). The excellent interfacial bonding between the KF-OX fibers and the PLA substrate allows for more efficient transfer of applied strain from the KF-OX fibers to the PLA substrate, resulting in increased tensile strength. However, when the content of KF-OX was further increased from 30% to 40%, the tensile strength of PLA nanocomposites decreased rapidly from 91.5 MPa to 53.6 MPa. This is due to the excess of entangled KF-OX fibers in the polymer matrix of PLA nanocomposites, which may hinder the recrystallization of PLA chains and weaken the stiffness of PLA nanocomposites (Chen et al.).
Wootthikanokkhan et al. investigated the effect of sisal fibers, Cloisite 30B nanoclay and hexagonal boronitrile (h-BN) fillers on the properties of PLA composites. They found that the addition of 5% kenaf fiber, Cloisite 30B nanoclay and h-BN all slightly improved the tensile modulus of PLA composites before and after annealing treatment. However, the addition of red hemp fibers increased the tensile modulus of PLA composites less compared to Cloisite 30B nanoclay or h-BN. This also suggests that Cloisite 3B nanoclay and h-BN can better improve the tensile modulus of PLA composites because of the good compatibility between Cloisite 30B nanoclay and h-BN fillers and PLA matrix (Wootthikanokkhan et al.). On the one hand, the hydroxyl functional groups on the alkyl ammonium surfactant-modified Cloisite 30B nanoclay formed polar interactions with the carbonyl functional groups of PLA chains by promoting the exfoliation of Cloisite 30B interlayer particles in the PLA matrix, which contributed to the increase of the tensile modulus of the PLA composites. On the other hand, the good compatibility between the h-BN filler and the PLA matrix is mainly attributed to the interaction between the carbonyl functional group of PLA and the lent electron pair of nitrogen on the surface of the h-BN filler, which can be confirmed by the bending vibration of B-N-B at 1360 cm-1 by IR spectroscopy. However, the lack of polar interactions between the sisal fibers and PLA chains resulted in poor interfacial adhesion between the sisal fibers and the PLA matrix, leading to reduced stiffness of the PLA composites.
Swaroop and Shukla investigated the effect of adding magnesium oxide particles (magnesium nanoparticles) on the properties of PLA nanocomposites. In their work, it was found that if the nano-MgO content was increased to 2% (mass fraction), it would gradually increase the tensile strength and elastic modulus of PLA nanocomposites from 29.1 MPa and 1.89 GPa to 37.5 MPa and 2.47 GPa, respectively. This is due to the fact that decreasing the size of MgO nanoparticles can increase the surface area/volume ratio of MgO particles, thus increasing the interfacial area of MgO nanoparticles. The addition of MgO nanoparticles to the PLA matrix can provide higher interfacial forces between the MgO filler and the PLA matrix, thus promoting the transfer of applied stress from the PLA matrix to the MgO nanoparticles and improving the mechanical properties of PLA. However, the opposite phenomenon was found when the loading level of nano MgO was increased from 2% to 4%. According to Swaroop and Shukla, the tensile strength and elastic modulus of PLA nanocomposites gradually decreased from 37.5 MPa and 2.47 GPa to 26.2 MPa and 1.96 GPa, respectively. This is attributed to the fact that the higher content of nano-MgO self-agglomerates into larger agglomerated particles, which weakens the interfacial adhesion between the agglomerated nano-MgO particles and the PLA matrix. As a result, the agglomerated particles achieve phase separation from the PLA matrix and act as stress concentration points in the PLA matrix, reducing the enhancement effect of nano-MgO in the PLA matrix.
8.3 Increase heat deflection temperature and heat resistance
The ability of a polymeric material to maintain its important properties for a period of time at maximum operating temperature is defined as heat resistance. The thermal behavior of polymer matrix is highly dependent on the crystallization behavior and crystallinity of the polymer material (Ma et al.). The chain segments of semi-crystalline PLA have been found to coexist in three forms: crystalline part, rigid amorphous part and flexible amorphous part (Nagarajan et al.). In the crystalline part of semi-crystalline PLA, the chain segments are arranged in an ordered crystalline structure. The crystalline chains coexist with the random long molecular chains in the amorphous part. When the temperature of PLA polymers reaches their glass transition temperature Tg, the PLA molecular chains in the crystalline region are less likely to move due to the presence of intermolecular bonds, while those in the amorphous region tend to move freely (Nagarajan et al.). There are some rigid PLA chain segments within the amorphous region that may impede the movement of the entire long molecular chain, i.e., the rigid amorphous portion. On the other hand, the remaining chain segments in the amorphous region with high activity at Tg are referred to as the movable amorphous portion. Due to the presence of a random arrangement of long molecular chains, the movable amorphous part has very low heat resistance at deformation temperatures close to Tg. In addition, heat resistance is usually evaluated by testing the softening point under a fixed load. Two measurement techniques are used to quantify thermal resistance: measurement of the Vicat softening temperature (VST) and measurement of the heat deflection temperature (HDT). The temperature at which a flat-tipped needle with a cross-sectional area of 1 mm2 pierces the sample to a depth of 1 mm is called VST. HDT is defined as the temperature at which the sample is deformed to 250 μm at a certain load (typically 0.46 MPa or 1.80 MPa) and thickness at a rate of 2 °C/min. To improve the heat resistance of PLA, nucleating agents can be added, PLA can be blended with heat resistant polymers, and PLA composites can be blended with nano-reinforced fillers and natural fibers.
8.3.1 Addition of nucleating agents
As mentioned earlier, the heat resistance of PLA is influenced by the crystallization behavior of the PLA matrix. The addition of nucleating agents can significantly change the crystallization behavior of PLA matrix, which has a significant effect on the heat resistance of PLA. In a study by Yu et al., they investigated the effect of increasing the concentration of coupling agent-modified talc on the mechanical and thermal properties of PLA composites. They found that increasing the talc content to 30% (mass fraction) significantly increased the glass transition temperature Tg and crystallinity Xc of the PLA composites. In addition, it was found that the increased talc content slightly decreased the cold crystallization temperature Tcc of PLA (Yu et al.). The significant effect of talc content on Tg, Tcc and Xc also indicated that the talc filler has a strong nucleation effect and can improve the crystallization ability of PLA. The increase in Tg is mainly attributed to the strong interfacial adhesion between PLA and talc particles, which can reduce the free volume in the PLA matrix and limit the movement of PLA chains. In other words, the higher Tg also indicates that the chains of disordered PLA molecules need to reach higher temperatures to move freely in the flowing amorphous fraction, thus enhancing the heat resistance of PLA. In addition, as shown in Table 8-3, the higher crystallinity also indicates that increasing the molecular bonding between PLA chains in the matrix helps to increase the crystallinity of the PLA matrix. On the other hand, it was reported by Yu et al. that the melting temperature Tm slightly decreased with the increase of talc content in the PLA matrix. This also indicates that the crystals nucleated on the surface of talc particles are usually imperfect and tend to break and melt at lower temperatures. In addition, it was observed that the HDT of PLA composites gradually increased with the increase of talc content until the talc content reached 30%. The addition of talc can increase the service temperature of PLA composites and make them withstand greater loads at high temperatures, thus improving the heat resistance of PLA composites.
Quchiar et al. also investigated the effect of clay minerals, talc and kaolinite as nucleating agents on the thermal properties of PLA composites. They found that increasing the content of talc and kaolinite, the glass transition temperature Tg of PLA composites remained around 57°C. It was also observed that the cold crystallization temperature Tcc of the PLA composites gradually decreased with increasing talc content. the decrease in Tcc was attributed to the fact that the PLA composites with the addition of talc had crystallized during the cooling process. However, they also observed that the addition of kaolinite had no significant effect on the thermal performance of PLA composites. Balakrishnan et al. found that there was no crystallization peak during cooling of PLA composites when MMT was added as a nucleating agent. This is believed to be due to the very slow crystallization rate of PLA during the cooling process. The decrease in Tg after the addition of MMT to the PLA matrix is mainly due to the plasticizing effect of the surfactant MMT. The alkylammonium surfactant used to modify the MMT particles can promote the intercalation effect of PLA molecular chains inserted into the interlayer gaps of MMT particles. Thus, this further leads to free movement of PLA molecular chains in the mobile amorphous fraction of the PLA matrix at lower Tg. They also observed that increasing the MMT content significantly decreased the crystallization temperature Tc. They also suggested that the presence of MMT particles could act as a nucleating agent by promoting the initial crystallization of PLA substrates. Wang et al. investigated the effect of increasing the CNT content on the crystallization and thermal properties of PLA nanocomposites. Increasing the CNT content significantly enhanced the crystallinity of PLA, which could increase the crystallinity from 4.7% to 12.46%. This may be attributed to the interfacial adhesion between CNT particles and PLA matrix due to the higher aspect ratio and larger specific surface area of CNT particles. In addition, the strong interfacial interactions between the -OH and -COOH groups of CNT particles and the PLA matrix can induce the formation of many nuclei, thus improving the crystallinity of the PLA matrix. They also found that the crystallization temperature Tc of all the PLA nanocomposites with CNT addition (Tc about 59 °C) was slightly lower than that of the original PLA (Tc = 59.7 °C). This also indicates that the presence of CNTs as nucleating agents promoted the initial crystallization of PLA. On the other hand, Zhou et al. investigated the effect of adding CNT with carboxyl group (CNT-COOH) as a nucleating agent on the thermal properties of PLA composites. They found that increasing the content of CNT-COOH significantly increased the initial degradation temperature and glass transition temperature of PLA/CNT- COOH nanocomposites. The increase in the initial degradation temperature and glass transition temperature may be due to the formation of strong interfacial bonds between CNT-COOH and PLA matrix, which hinders the movement of PLA random molecular chains.
According to Tang et al. , the addition of ethylenebis-hydroxystearamide (EBH) as a crystal nucleating agent and annealing at a melt crystallization temperature of 105°C for 20 min rapidly increased the crystallinity of PLA from 19% to 42%. This is due to the fact that part of the molten amorphous region in the PLA undergoes a continuous recrystallization process, resulting in a more perfect and thicker wafer. In addition, the addition of EBH rapidly increased the HDT of pure PLA from 51% to 93%. The HDT of PLA composites with EBH addition started to increase when the crystallinity of PLA increased more than 20%. In addition, they found that the significant increase in HDT of PLA composites was mainly due to the increase in PLA crystallinity.
8.3.2 Poly(lactic acid) blended with heat resistant polymers
Poly(ε-caprolactone) (PCL) and poly(ε-caprolactone/L-propyleneglyceride), P(CL/L-LA) blends with poly(L-lactide) (PLLA) significantly lowered the glass transition temperature Tg and melting temperature Tm of PLLA. The lower Tg of PLLA blends indicated that PCL/PLLA blends and P(CL/L-LA) blends promoted the crystallization of the polymer matrix at lower temperatures. The lower Tg of the PLLA blends indicates that the PCL/PLLA blends and P(CL/L-LA) blends promote the crystallization of the polymer matrix at lower temperatures, thus improving the crystallinity of the PLLA blends. In addition, they found only one Tg when PLLA was mixed with P(CL/LLA), indicating that P(CL/L-LLA) is mutually soluble with PLLA. However, no Tg was found when copolymerized with P(CL/L-LLA), and Tg disappeared from the differential scanning calorimetry (DSC) curve. However, PCL was found to be incompatible with PLLA due to the fact that their blends have two melting temperatures. In a study conducted by Cock et al., DSC analysis showed that the blending of PCL with PLA could rapidly increase the crystallization rate of PLA blends. In addition, since the melting temperature of PCL is about 55 °C, the PCL blending with PLA also shifts the Tg of the original PLA to a lower value. This also reduces the thermal crystallization of PLA by supporting the nucleation of PCL during the post-melting of the PLA matrix. D'Amico et al. found that poly(3-hydroxybutyrate) (PHB) blends with PLA improved the heat resistance of PLA by increasing the crystallization rate of PLA. Increasing the content of PHB in the PLA matrix rapidly reduced the Tg of the PLA blends by promoting earlier crystallization of the PLA/PCL matrix at lower Tg. In addition, up to 70% (mass fraction) of PHB blended with PLA also increased the crystallinity from 2.7% to 35.2% of pure PLA. This is due to the fact that PHB is co-blended with PLA, which increases the crystalline fraction and thus decreases the proportion of amorphous flowable fraction and ultimately promotes the formation of crystals in the PLA matrix.
This is due to the fact that PHB is co-blended with PLA, which increases the crystalline fraction and thus decreases the proportion of amorphous flowable fraction and ultimately promotes the formation of crystals in the PLA matrix. This is due to the constraining effect of a large number of small PA grains. Hashima et al. (2010) reported that increasing the PC content significantly increased the HDT of styrene-butadiene-styrene block copolymers blended with PLA due to the higher crystallinity and Tg of polycarbonate (PC). On the other hand, Guo et al. found that mixing 50% (mass fraction) of polyformaldehyde (POM), a polymer with high heat resistance properties, greatly increased the HDT of PLA from 65°C to 133°C. In addition, the Tg of PLA/POM blends slightly decreased with the increase of POM content, which indicated that the presence of POM could promote the early crystallization process of PLA.
8.3.3 Poly(lactic acid) composites blended with natural nanofibers
The addition of natural fibers such as sisal and wood fibers to PLA to improve the properties of PLA, including heat resistance, has attracted the interest of many researchers. According to Renstad et al., blending 20% of kenaf fibers significantly increased the HDT of annealed PLA from 99.7°C to 128°C. The increase in the HDT value of PLA may be due to the isothermal crystallization of PLA facilitated by the fiber shape of kenaf fibers with high aspect ratio. This finding was further confirmed by the increase in PLA crystallinity from 33.9% to 374% when 20% of kenaf fiber was added (Renstad et al.). On the other hand, Huda et al. found that adding 40% of untreated kenaf fibers to PLA composites at an applied load of 0.46 MPa and a heating rate of 2°C/ min greatly increased their HDT values from 64.5°C-1.2°C to 170.3°C-1.0°C. In addition, they investigated the effects of silane and alkali treatments on the HDT of PLA composites. The silane-treated sisal fiber-reinforced PLA composites showed the highest HDT value (174.8°C ± 1.1°C), which was greater than that of other materials, such as untreated sisal fiber-reinforced PLA composites (170.3°C ± 1.0°C), alkali-treated sisal fiber-reinforced PLA composites (172.8°C ± 0.9°C), and alkali-silane treated sisal fiber-reinforced PLA composites ( 173.4°C±1.0°C). It is possible that the heat resistance of all sisal fiber-reinforced PLA composites was greatly improved due to the ability of sisal fibers to limit the deformation of sisal fiber-reinforced PLA composites, thereby promoting the crystallization of the PLA phase in the composites. In addition, the high aspect ratio of long kenaf fibers can improve the interfacial bonding between PLA matrix and kenaf fibers by increasing the interfacial bonding strength.
Awal et al. investigated the effect of adding wood pulp and wood pulp/bioamides to PLA on the thermal behavior of PLA biocomposites by thermogravimetric analysis and HDT tests. They reported that the temperature of virgin PLA at 5% weight loss (mass fraction) (T5%) was 304°C, while the temperature of T5% for wood pulp was 208°C. T5% was found to occur at 274°C for both wood pulp-reinforced PLA biocomposites and wood pulp/biomamide reinforced PLA biocomposites. Adding wood pulp and wood pulp/biodiamide to PLA, the temperature at maximum weight loss of the original PLA increased significantly from 350°C to 450°C. On the other hand, the addition of wood pulp to PLA slightly increases the HDT of the original PLA from 54°C to 56°C. The incorporation of bioimide in wood pulp increased the HDT of PLA biocomposites to 61°C significantly. This suggests that the presence of biomimic improves the wetting of pulp fibers in the PLA matrix and enhances the interfacial adhesion strength of pulp fibers to the PLA matrix (Awal et al.). Therefore, the addition of bioamides to wood pulp can increase the temperature of PLA composites for use in materials engineering.
8.4 Flow enhancement, melt strength, rapid prototyping time
The melt strength of a polymer is one of the most important properties used to determine the processability of a particular polymer, especially in applications with high melt stretch or tensile flow (e.g., film and mold molding). In industrial applications, the low melt strength of PLA may lead to poor processability and product quality, such as the presence of unstable bubbles during blown film and sinkage during blow molding (Liu et al.; Field et al.). In order to improve the melt strength and processability of PLA, various modification methods have been intensively researched and developed, such as the addition of crosslinkers, plasticizers, chain extenders and blending with polymers with higher melt strength (Liu et al.).
8.4.1 Cross-linking agents
The conventional route of lactic acid polymerization cannot produce high molecular weight PLA in a limited polymerization time. the low molecular weight of PLA results in poor melt strength and processing properties, which may lead to a decrease in the quality of the final PLA product. To improve the melt strength of PLA, suitable cross-linking agents can be introduced during the processing of PLA blends or high-energy electron methods can be used. Nijenhuis et al. prepared cross-linked PLA by adding a cross-linking agent of diisopropyl peroxide at a higher curing temperature to improve the melt strength of PLA. In addition, other researchers (Liu et al.; Yu et al.) used Luperox and lauryl peroxide as cross-linking agents to improve the melt strength and melt viscosity of PLA by inducing cross-linked structures. Liu et al. showed that the addition of Luperox and lauryl peroxide significantly improved the melt strength of PLA, but the presence of the cross-linked structure limited the slippage of the PLA melt, resulting in lower melt strain. They also found that macromolecular size (Mz) is related to melt strength and flexural lifetime because intermolecular interactions limit the deformation of PLA substrates. Yu et al. also improved the melt strength and processing properties of PLA by adding 0.3% Luperox and 0.3% lauryl peroxide to PLA, respectively. The addition of 0.3% L-uperox increased the melt strength of the original PLA from (10.5±2.1)×10-2 to (18.3±1.4)×10-2N . In contrast, the addition of 0.3% lauryl peroxide resulted in a melt strength of PLA of (15.1 ± 2.1) × 10-2 N. This was due to the cross-linking reaction that caused the formation of long branched chains, enhanced the elongation fluid tensile hardening phenomenon, and limited the flowability of PLA. As a result, the melt strength and melt viscosity of the cross-linked PLA were significantly increased. On the other hand, increasing the content of the cross-linking agent benzoyl peroxide (BPO) to 1% (mass fraction) significantly improved the melt strength and melt viscosity of PLA by promoting the formation of a three-dimensional network (Zhang et al.).
8.4.2 Chain extender
The use of various chain extenders such as diisocyanates and 1,4-butanediol with PLA can improve the melt strength and processing properties of PLA by increasing its molecular weight (Liu et al.). Di et al. modified PLA with chain extenders (1,4-butanediol and 1,4-butanediisocyanate) to improve the melt strength, melt viscosity and processing properties of PLA. The addition of 1,4-butanediol and 1,4-butanediisocyanate significantly improves the melt strength and melt viscosity of the modified PLA, allowing the production of PLA foams with smaller pore size and larger pore density. On the other hand, Liu et al. and Yu et al. also investigated the effects of chain extenders (homophthalic dianhydride or PMDA and oxazoline) on PLA melt strength, melt viscosity and processing properties. The addition of PMDA and oxazoline significantly increased the melt strength, melt rate, and melt viscosity. The increase in melt strength, melt rate and melt viscosity mainly corresponds to the increase in molecular weight such as Mz, whose larger intermolecular bonding limits the deformability of the chains (entanglement occurs), resulting in a higher melt strength. Importantly, this observation was also reported by Yu et al.
8.4.3 Blending with other polymers
Polymer blending is one of the most common methods to improve the melt strength of polymers at low melt strengths and poor processability. In the work of Liu et al. and Yu et al., the blending of PLA with the biodegradable polyesters Bionolle (copolymer of polybutylene succinate) and Ecoflex (aliphatic aromatic copolymer) slightly improved the melt strength and processing properties. The enhancement of melt strength and processing properties by Bionolle and Ecoflex is mainly attributed to the partial compatibility of these polymers with PLA, which increases the physical interaction between molecules in the polymer blend and the deformation resistance of the polymer chains. Moreover, the good compatibility of PLA and Biomax led to the significant improvement of melt strength, melt strain and processing properties by the blending of PLA with Biomax (elastomer), which induced intermolecular interactions in the polymer blend system (Liu et al.). According to Gu et al., the blending of PLA with polybutylene adipate-terephthalate (PBAT) improves the melt strength, viscosity, and processability because the blended PLA/ PBAT melt has a stronger tendency for complex shear thinning. Zhang et al. blended cross-linked PLA (cross-linked with 0-1% BPO) with PBAT to further improve the melt strength, viscosity, and processing properties. Their results showed that the addition of PBAT can effectively improve the melt properties of cross-linked PLA such as melt strength and viscosity through chain entanglement, thus resisting the deformation of polymer chains (Zhang et al.).
8.4.4 Plasticizers
Plasticizers are often added to polymer blends to improve their processing properties. For PLA, the addition of plasticizers can effectively lower the glass transition temperature Tg and reduce the melting temperature and crystallinity of the semi-crystalline polymer PLA (Farah et al). Much research has also been done on the use of various plasticizers, such as polyethylene glycol (PEG), acetyltri-n-butyl citrate (ATBC), glucose monoesters, meta-fatty acid esters, and epoxidized soybean oil (ESO) to improve the processing properties as well as the toughness of PLA (Arrieta et al.; Jacobsenm, Fritz; Xu, Qu; Liu et al.). Xu and Qu showed that the melt strength of PLA increased gradually with the increase of ESO content of plasticizer and reached a maximum at 6% (mass fraction) of ESO. The increase in melt strength is due to the long branched molecules of PLA that increase the intermolecular interaction forces in the PLA melt, thus limiting the deformation of the polymer chains. In addition, the interaction between ESO and PLA may also contribute to the formation of hydrogen bonds between the epoxy group of ESO and the carbonyl group in the ester bond of PLA. Tee et al. investigated the effect of adding two plasticizers, epoxidized palm oil (EPO) and ESO, on improving the properties and processability of PLA. It was found that the addition of EPO and ESO significantly reduced the torque (maximum and final values), the material temperature and the time required to reach a homogeneous mixture. This suggests that the combination of EPO and ESO can provide better lubrication and thus improve the processing performance of PLA. EPO plasticizers have been found to be less viscous than ESO and have better results in assisting PLA processing.
Li et al. synthesized the plasticizer glycerol lactate with the aim of improving the thermal processing and melting properties of PLA/PVA blends. Due to the presence of both lactate and hydroxyl groups on glycerol lactate, its addition improves the processing properties of PLA/PVA by enhancing the compatibility between PLA and PVA. Maiza et al. investigated the effect of adding two different plasticizers, triethyl citrate (TEC) and ATBC, to PLA. The addition of TEC and ATBC reduces the Tg and melt viscosity of PLA and improves the processability of PLA. This is due to the low molecular weight of TEC and ATBC, which allows the TEC and ATBC molecules to occupy the space between the polymer chains and increase the mobility of the PLA chains. Saravana et al. investigated the effect of polyethylene glycol 1500 (PEG1500) and polyethylene glycol 6000 (PEG6000) grades of plasticizers on the properties of talc-reinforced PLA composites. The Tg of the talc-reinforced PLA composites increased with the increase of PEG1500 and PEG6000. This indicates that PLA has good compatibility with these plasticizers, which can enhance the fluidity of PLA chains and thus improve their processing properties. In addition, the simultaneous addition of PEG1500 and PEG6000 can improve the processing performance by overcoming the agglomeration of talc in the PLA matrix.
8.5 Special additives: antistatic agents, impact modifiers, fiber bulking agents / coupling agents
8.5.1 Antistatic agents
For hydrophobic polymers, such as PLA, discharge during the molding process can attract dust or dirt and cause office equipment to malfunction. To overcome this problem, it is often possible to prevent the formation of static electricity on the surface of these polymers by adding antistatic agents. The effectiveness of antistatic agents is measured by measuring the surface resistivity of polymer blends (Niaounakis). The addition of antistatic agents to hydrophobic polymers such as PLA can significantly reduce the surface resistivity of the polymer by 109 to 1012 Ω (Niaounakis). In order to reduce the surface resistivity or rather to increase the electrical conductivity of polymers, conductive fillers such as carbon black, CNT, carbon fibers, conductive ceramics and powdered metals must be added to hydrophobic polymers (Moon et al.; Silva et al.). According to Silva et al., when the addition of carbon black reaches 15%, it will significantly increase the conductivity and decrease the resistivity of PLA composites. These properties allow PLA composites with added carbon black to be used as antistatic packaging for the transportation and storage of electronic devices (Silva et al.). On the other hand, the addition of CNT with semiconducting behavior can improve the electrical conductivity of polymer composites as described by Moniruzzaman and Winey. The addition of CNT to PLA matrix can significantly reduce the surface resistivity of PLA composites and improve the efficiency of electromagnetic wave shielding (Moon et al.).
8.5.2 Impact modifier
As mentioned earlier, PLA can be processed using conventional melt processing techniques such as extrusion and injection molding. In addition, it has good biocompatibility, biodegradability and tensile properties. However, the poor toughness (high brittleness) and low ductility of PLA limit the use of PLA in various applications. To improve the toughness of PLA, impact modifiers can be used in PLA blending systems to reduce the brittleness of PLA without weakening its stiffness. Several companies have introduced impact modifiers that are designed for PLA applications. Notta-Cuvier et al. added 10% (mass fraction) of Biomax Strong (BS) 100 impact modifier to PLA, which significantly improved the elongation at break and tensile properties of PLA. In addition, the addition of 10% (mass fraction) BS to the plasticized Cloisite 25A/PLA composite resulted in good ductility while maintaining the stiffness and strength of the PLA composite. Mat Taib et al. increased the content of BS impact modifier to 50% (mass fraction), which significantly improved the notched impact strength and elongation at break of PLA. This also indicates that the addition of BS impact modifier can improve the toughness of PLA. However, the tensile modulus and yield stress of PLA were found to decrease with the increase of BS impact modifier. This is due to the fact that the toughening effect of BS impact modifiers reduces the crystallinity of PLA by increasing the plastic deformability of the PLA matrix. Barletta et al. added 1.8% (mass fraction) of Paraloid BPM-515 impact modifier to PLA/talc composites to improve the compatibility effect between PLA and talc filler to improve the toughness. In a study conducted by Diaz et al., the addition of Paraloid BPM-515 impact modifier to PLA slightly increased the elongation at break. In addition, it was observed that the impact strength of PLA increased rapidly with the addition of Paralois BPM-515 impact modifier, which also indicates the toughening effect of PLA. Choochottiros and Chin studied and synthesized two different transparent impact modifiers for PLA. They synthesized poly(butadiene-methyl methacrylate-butyl methacrylate-butyl acrylate-methyl butyl acrylate-hydroxyethyl methacrylate) (called BMBH copolymer) and poly(butadiene-propylene-methacrylate-butyl methacrylate-butyl methacrylate) (called BLMB copolymer) as PLA impact modifiers, which can improve toughness and impact strength by increasing compatibility with the PLA matrix while maintaining transparency of PLA. They can improve toughness and impact strength by increasing compatibility with the PLA matrix, while maintaining the transparency of PLA. However, as mentioned previously, all commercially available PLA impact modifiers are non-biodegradable and are typically used in industrial applications in 10% (mass fraction) amounts (Notta-Cuvier et al.). Since the previously mentioned commercially available impact modifiers are not biodegradable, various types of biodegradable polymers are used as biodegradable impact modifiers for PLA. PCL is one of the most commonly used biodegradable polymers used as impact modifiers for PLA (Wang et al.; Broz et al.; Notta-Cuvier et al). According to Wang et al., the reaction-compatible PLA/PCL blends achieved synergistic compatibility and toughening when the PLA/PCL mass ratio was 80/20. There are also biodegradable polymers that have been used as degradable impact modifiers for PLA, such as polypropylene carbonate, polybutylene succinate, poly(p-dioxanone), polybutylene adipate-terephthalate terephthalate, and poly(tetramethylene adipate-terephthalate) (Notta-Cuvier et al). Odent et al. investigated the application of poly(ε-caprolactone-co-δ-pentyl lactone) (a random aliphatic copolyester) as a biodegradable impact modifier, which improved the toughness of PLA while maintaining the transparency of PLA.
8.5.3 Fiber bulking agent/coupling agent
There are many scholars who have studied the enhancement of the mechanical properties of natural fibers (e.g., kenaf and flax fibers) on degradable polymers such as PLA and polyvinyl alcohol (John, Anandjiwala; Foruzanmehr et al.; Lee et a1). Natural fibers with biodegradability, high toughness, high specific strength, low cost, low density, and renewable are ideal for replacing traditional reinforcing fillers such as calcium carbonate in PLA composites (Foruzanmehr et al.). However, the low compatibility between hydrophobic PLA and hydrophilic natural fibers significantly weakens the mechanical-physical properties of PLA composites and limits the application of PLA composites (Kumar et al.; Lee et al.). A study conducted by Foruza- nmehr et al. showed that the interfacial adhesion between flax fibers and PLA substrates could be significantly improved by coating the flax fiber surface with an oxidized TiO2 film using a solution-gel dip coating technique. A study conducted by Foruza- nmehr et al. showed that the interfacial adhesion between flax fibers and PLA substrates could be significantly improved by coating the flax fiber surface with an oxidized TiO2 film using a solution-gel dip coating technique. This is mainly due to the good interfacial bonding between the fiber and PLA matrix by effectively transferring the stress between the fiber and the PLA matrix. Lee et al. investigated the effect of 3-epoxypropyltrimethoxysilane as a coupling agent on the mechanical properties of sisal fiber PLA composites. Treatment of kenaf fibers with 3-epoxypropyltrimethoxysilane significantly improved the interaction between kenaf fibers and PLA substrates (Lee et al.). Wang et al. investigated the effect of using four different coupling agents, vinyltrimethoxysilane (vinyl silane), γ-aminopropyltriethoxysilane (amino silane), γ-epoxypropyltrimethoxysilane (epoxy silane) and γ-methacryloxypropyltrimethoxysilane (allyl ester silane), along with the addition of wood flour, on the mechanical properties of wood flour/PLA The effect of mechanical properties of the composites. The addition of aminosilanes, epoxy silanes and allyl ester silanes significantly improved the tensile strength, elongation at break and impact strength of the wood flour/PLA composites. This is because the silane coupling agent forms a "bridge" between the PLA matrix and the wood powder, thus improving the interfacial interaction between the PLA matrix and the wood fiber.
8.6 Conclusion
The undesirable properties of PLA, such as brittleness, low toughness, low thermal stability and poor processability, can be improved by adding various additives, including reinforcing fillers. In order to expand the application scope of PLA and improve the existing properties of PLA, various suitable additives have been investigated by different scholars. In addition, a variety of reinforcing additives have been used to improve the mechanical properties of PLA to achieve the target performance required by various industries. The addition of processing aids to PLA blends is also intended to improve the processing properties of PLA during melt processing. Typically, additives and processing aids are added to improve the mechanical, processing, thermal and other specific properties of PLA while maintaining its biodegradability and biocompatibility.
9. PLA molding process
9.1 Extrusion
The extrusion process of polylactic acid (PLA) consists of three main stages: continuous melting of PLA, conveying and extrusion through the mouth die. PLA materials can be melt-processed and blended using an extruder to make PLA compounds, which are then processed into final products by blow molding, injection molding, and thermoforming processes (Lim et al.). A typical screw extruder is divided into three main sections, the feed section, the transition section and the metering section. For the feed section, which receives the PLA compound particles and delivers them to the screw, and for the transition section, the thread depth of the screw is designed to gradually decrease to squeeze the PLA particles, thereby increasing the contact between the PLA particles and the machine jig (Lim et al.). The transition section is also referred to as the compression section. For the metering section, the slot depth of the screw is constant and shallow in order to pump the exact amount of molten PLA (Kuhnert et al.; Lim et al.). L/D and compression ratio are the most important screw parameters of an extruder. l/D is the ratio of screw thread length to screw outside diameter and is used to determine the shear and residence time of the PLA melt. A screw with high L/D helps to provide better mixing, shear heat and longer melting time (Lim et al.). Conversely, the compression ratio is the ratio of the slot depth in the feed section to the slot depth in the metering section. The compression ratio is used to evaluate the shear heat provided by the screw. In other words, the higher the compression ratio of the screw, the higher the shear heat provided to the PLA melt. Typically, for PLA processing, a typical screw with an L/D of 24 to 30 and a compression ratio range of 2 to 3 is recommended (Kuhnert et al.).
In the PLA plasticization process, PLA resin (pellet or powder form) enters the screw groove of the extruder from the feed hopper (Lim et al.) and the compressed PLA resin (pellet or powder form) is conveyed to the end of the screw groove using a screw in the screw groove driven by a hydraulic or electric motor. Heat is provided by a heater wrapped around the screw channel to melt the compacted PLA resin. The screw groove on the screw shears and compacts the PLA resin into the screw channel. During the passage of the compacted PLA through the transition zone, friction due to contact between the PLA resin and the screw groove generates additional heat to melt the PLA resin. The combination of heat from the heating sheet and frictional heat due to the shear between the PLA resin and the screw and screw groove walls can provide enough heat to bring the compressed PLA to the melting point of PLA (in the range of 170 to 180°C) before it reaches the end of the screw groove (Lim et al.). The temperature of the heating sheet wrapped around the spiral groove is typically set in the range of 200 to 210°C to ensure that all crystalline phases in the PLA are melted and the optimum melt processing viscosity is obtained. After extrusion from the transition zone, the PLA melt enters the metering zone. In the metering zone, the pump generates enough pressure to push the PLA melt out of the mouth die.
9.2 Blow molding
Nowadays, due to increasing environmental awareness, biodegradable polymers such as LA have been used in some food and beverage industries instead of non-biodegradable polymers (e.g. polypropylene, polyethylene, etc.) for the preparation of beverage bottles. However, the use of PLA bottles in the beverage industry is limited to beverages that are not sensitive to oxygen, such as pasteurized milk and pure water beverages (Lim et al.). Various technologies including internal plasma deposition, multilayer PLA bottles, and external coatings have been used to form barriers within PLA and between beverages. However, the high production costs of these technologies have limited their application in the beverage industry.
Injection stretch blow molding technology is mainly used for the production of PLA-based bottles (Lim et al.). Basically, there are two methods for producing PLA bottles by injection stretch blow molding technology: one-step and two-step methods. The production of PLA bottles using a two-step process involves two sequential steps:an injection molding machine and a blow molding machine (Castro-Aguirre et al.). Initially, injection stretch blow molding requires the use of an injection molding machine to form the preforms (also called billet molding). Afterwards, the blanks are transferred to a blow molding machine. In the blow molding machine, the blanks are heated to 85 to 110°C by infrared heaters and blown in the circumferential direction while extending axially to achieve biaxial orientation. The production of biaxially oriented PLA bottles improves the physical properties and barrier properties of PLA bottles by generating strain-induced crystallization and stabilizing the amorphous zone in the PLA bottle. In addition, the power of the infrared heaters was set at different levels to find the optimal stretching process for stretching the molds into bottles with uniform wall thickness (Lim et al). Moreover, heat stabilizers (e.g., carbon black, etc.) were mixed with PLA to increase the absorption of infrared energy. PLA blanks tend to shrink during reheating, especially in the areas near the neck and end caps of the bottles produced, where the injection residual stresses are highest (Castro-Aguirre et al.). Designing preforms with gradual transition areas minimizes shrinkage problems. When the preforms reach the optimum temperature, they are transferred to the blow mold and then the nozzle is adjusted to seal the preforms. At the same time, the tension rod moves toward the preform at a speed of 1.0 to 1.5 m/s and stretches the preform to the bottom of the blow mold. When the stretching rod moves toward the preform, compressed air with a pressure of 0.5 to 2.0 MPa is slowly blown into the preform to prevent contact between the preform and the stretching rod. When the tension rod reaches the bottom, adjust the compressed air force to 3.8 to 4.0 MPa to completely fill the bottle so that the bottle takes the shape of the blow mold. Before removing the bottle from the mold, compressed air needs to be continuously blown into the bottle and held for a few seconds to cool the bottle.
On the other hand, injection stretch blow molding using the one-step process is done in the same step for both injection and blow molding production. In other words, the one-step process requires the integration of the injection molding unit and the blow molding unit on the same machine (Castro-Aguirre et al.). In the one-step process, the injection molded blanks are partially cooled to about 110°C and then further stretched by air blown through the blow molding unit. Comparing the one-step and two-step injection stretch blow molding processes, it is clear that the preforms produced in the two-step process need to be reheated after transfer to the blow molding machine, which can cause slight degradation of the PLA blanks, especially the neck, and make the PLA brittle. Therefore, different designs may be required for one-step and two-step production processes (Lim et al.).
PLA resin also exhibits strain hardening when stretched at high strain ratio. Therefore, the stretching process of PLA preforms should be optimized to obtain PLA bottles with optimal sidewall orientation and wall thickness. In addition, PLA bottle blanks must be designed to match the size and shape of the PLA bottle in order to obtain the optimum stretch ratio of the PLA bottle blanks during the blow molding process; under-stretching of PLA bottle blanks can result in large variations in the wall thickness and poor mechanical properties of PLA bottles. On the other hand, overstretching may lead to the formation of microcracks on the surface of PLA bottles, which diffracts light and causes stress whitening. In the application of PLA bottles, the recommended important key properties and major stretching ratios are: 2.8 to 3.2 for the axial stretching ratio, 2 to 3 for the circumferential stretching ratio, and 8 to 11 for the ideal flat stretching ratio (Lim et al.). In addition, some standard features of PLA bottle design such as bottle shape, transition shape, step changes, pinch points on the core and cavity need to be considered in order to obtain the best design of bottle blanks. The correct design of bottle blanks is important to produce PLA bottles with good mechanical properties, physical properties and transparency.
9. 3 Injection molding
Injection molding is one of the most widely used processing methods to transform polymers, including PLA, into thermoplastic finished products with complex shapes and high dimensional accuracy (Kuhnert et al.). In the injection molding machine, a three-stage screw extrusion unit is installed to plasticize the polymer melt. The extrusion unit in the injection molding machine is designed with a screw that reciprocates in the barrel to melt the polymer resin from the resin inlet using frictional heat and then presses the polymer melt into the mold cavity. The injection molding cycle consists of eight major stages. The closing of the mold is the beginning of the injection molding cycle. Immediately after the mold closes, the nozzle will open and the screw will move forward to inject the desired amount of polymer melt into the mold cavity. After the polymer melt is injected into the mold cavity, continuous holding pressure is applied to hold the screw in a forward position to compensate for the shrinkage of the polymer material during cooling. The cooling process of injection molding of polymer product parts exacerbates the shrinkage deformation of the material and is accompanied by the onset of crystallization and rapid increase in density, providing a cooling time long enough to stabilize the polymer product dimensions. The screw rotates and moves the polymer melt forward during the cooling of the injected polymer part. When the polymeric part has cooled sufficiently, the mold is opened and the injected polymeric part is unloaded (Lim et al.; Kuhnert et al.).
In order to maximize the productivity of the injection molding process, the cycle time is one of the most important parameters and is often required to be minimized. Usually, transferring partially cooled injection molded parts to a post-molding cooling unit can help reduce the cycle time of the injection molding process by reducing the cooling time and extending the cooling time of the injection molded parts outside the mold (Kuhnert et al.) In addition to this, it is possible to increase productivity by reducing the duration of non-processing steps in the injection molding cycle, such as the mold opening time, the part ejection time from the and mold closing times. Moreover, lowering the mold temperature also increases the rate of heat transfer out of the polymer (Kuhnert et al.). On the other hand, injection molding cycle is also important to control and reduce the shrinkage effect of PLA injected parts with brittle behavior due to accelerated aging of PLA. Also, the filling, holding and cooling phases in the injection molding cycle are important factors that lead to shrinkage of the injection molded polymer parts. The low crystallinity of injection molded PLA parts is mainly due to the slow crystallization rate of PLA, which leads to the inability to arrange the long chains of PLA in an orderly structure (Kuhnert et al.). In addition, process parameters such as cooling rate, mold temperature, holding pressure and post molding cooling treatment need to be precisely controlled to prevent shrinkage of the PLA injection molded parts.
On the other hand, the mechanical, physical and thermal properties of injection molded PLA parts depend heavily on the processing parameters during the injection molding process. These processing parameters include mold temperature, melt temperature, holding pressure, injection flow rate, maximum shear rate and shear stress. According to Kuhnert et al., the processing parameters of melt temperature and injection flow rate affect the shear rate and shear stress of the polymer melt injected into the cavity, as well as the thermal recovery strain test and crystallinity. Higher injection flow rates and lower melt temperatures contribute to higher shear rates and shear stresses in the molecular orientation, resulting in higher crystallinity. In addition, molecular orientation and crystallinity greatly affect the mechanical properties of injection molded PLA parts, such as tensile strength and elongation at break. As mentioned earlier, chain orientation affects the crystallinity of injection molded PLA parts due to shear stresses during the injection process. A technique known as "shear-controlled orientation" in injection molding can be used to enhance the mechanical properties of PLA (a semi-crystalline polymer). This technique can change the morphology of the polymer solute in the cavity in the cured state through push-pull effects and the ability to externally control in-mold shear and cooling (Castro-Aguirre et al.; Lim et al.)
9.4 Thermoforming
Thermoforming is a process that uses vacuum pressure to press pre-heated flexible plastic into a desired shape. Thermoforming is a common processing method for producing PLA packaging containers with simple characteristics, such as disposable cups, disposable food trays, lids and blister packs (Castro-Aguirre et al.). First, extruded PLA sheets thicker than 254 μm can be heated using an infrared radiation heater. The infrared red wavelength used to heat the PLA sheet must match the maximum absorbance of the PLA sheet so that the heating element is set at a temperature where most of the energy is absorbed by the PLA sheet. The heated PLA sheet is then thermoformed in a mold with a temperature range of 80 to 110°C. Aluminum molds are typically used to thermoform PLA packaging containers. In the case of thermoforming, the orientation of the PLA packaging containers can help increase the toughness (Lim et al.). Prior to the thermoforming process, the extruded PLA sheets are very brittle at room temperature. In order to avoid web breakage, the radius of the unfolding device and the skeleton rewinding device should be prevented from being too small. Thermoformed PLA parts are less brittle than flanged and lipped ones in areas that are highly stretched during the foaming process. The PLA extruded sheet should be heated to 90°C before trimming the PLA extruded sheet prior to thermoforming. In addition, PLA extruded sheets should not be exposed to temperatures higher than 40°C to prevent the sheets from unfolding due to the low heat deflection temperature of PLA sheets. PLA parts prepared by thermoforming should be stored at temperatures below 40°C to prevent the molecular weight decomposition of PLA when exposed to higher temperatures (Lim et al.).
9.5 Advanced technologies: 3D PLA printed, electron beam irradiation
9.5.1 3D Printing
See Chapter 10, Section 10.4 for more information on 3D printing.
9.5.2 Electron beam irradiation
Radiation crosslinking is one of the most widely used techniques to enhance the properties of polymers. Typically, various forms of high-energy radiation, such as electron beams, X-rays and γ-rays, can optimize the properties of polymers by forming three-dimensional networks (Murray et al.). Electron beam irradiation technology is one of the most effective methods to form cross-linked networks in polymer matrix by adding cross-linking agents. The application of electron beam irradiation methods can be used to enhance the properties of polymers. When a polymer is exposed to electron beam radiation, the electrons released by the electron beam gas pedal attack the polymer's polymer chains, resulting in the formation of polymer radicals. These high-energy polymer radicals tend to react in the polymer matrix to form cross-linked networks (C-C intermolecular bonding). Due to two important mechanisms - cross-linking and chain-breaking reactions - the application of electron beam irradiation can modify the structure and mechanical properties of the polymer matrix. During irradiation, cross-linking reactions and chain segment reactions occur simultaneously in the polymer matrix. If the cross-linking reaction predominates over the chain-breaking reaction during irradiation, the molecular weight of the cross-linked polymer will increase significantly. The dominant cross-linking reaction can significantly improve the physical and mechanical properties of the polymer (Rytlewski et al.; Ng et al.). On the contrary, if there are more chain-breaking reactions than cross-linking reactions, it will lead to severe degradation of the polymer matrix, and this will cause a decrease in the molecular weight of the polymer. During polymer irradiation, a suitable crosslinking promoter needs to be added in order to make the crosslinking reaction superior to the chain-breaking reaction of the chain segments. According to Ng et al., when irradiated with higher doses of electron beam radiation, the pristine PLA tends to have a chain-breaking reaction rather than a cross-linking reaction. This is because the high energy of electron beam irradiation can attack the PLA backbone by disrupting the building between carbon atoms on the PLA backbone, resulting in more random short radicals. These random short radicals react with oxygen in the air to form peroxyl radicals. As a result, the process of chain hydroperoxidation in PLA is initiated by further extending the chain-breaking preaction in PLA (Loo et al). The predominant chain-breaking reaction significantly weakened the mechanical properties of PLA and made it brittle. The addition of the cross-linking promoter trimethylpropane trimethacrylate (TMPTMA) to PLA significantly promoted the cross-linking reaction under irradiation conditions and formed a three-dimensional network in the PLA matrix. This is because the presence of TMPTMA can generate more monomeric radicals and form a large number of cross-linked networks in the PLA matrix.
In contrast, blends of high radiation resistant polymers such as low density polyethylene (LDPE) with PLA were also shown to promote the predominant cross-linking reaction in the PLA blend matrix rather than the chain breaking reaction. Bee et al., found that blending LDPE with PLA prolonged and promoted the cross-linking reaction in the polymer matrix LDPE / PLA blend. It was also found that when the LDPE/PLA matrix was irradiated by electron beam, the degree of cross-linking formed in the LDPE/PLA matrix was increased with increasing LDPE content. This is due to the fact that the application of electron beam irradiation tends to release more long-chain polymerization radicals, thus enhancing the formation of molecular weight crosslinking networks in the LDPE/PLA matrix. The mechanical properties of electron beam irradiated LDPE/PLA blends can also be improved by the addition of kenaf fibers. According to Bee et al., the elongation at break of LDPE/PLA blends decreased with increasing loading of kenaf fibers and increasing irradiation dose. This is attributed to the poor interfacial bonding between the sisal fibers and the polymer matrix of LDPE/PLA, which severely limits the migration of polymer radicals in the polymer matrix. As a result, the degree of cross-linking in the polymer matrix is reduced and the slip effect of the polymer chains in the LDPE/PLA matrix is limited.
9.6 Recycling methods and sustainability of PLA and its composites
Although PLA is a biodegradable polymer with the identification code shown in Figure 9-1, it still has many similarities to petroleum-based polymers. After use, PLA can also be physically broken, melted and processed into different products. In other words, PLA has the properties of a thermoplastic and can be recycled using the melt reprocessing cycle. European Bioplastics (European Bioplastics, 2019) reports that the feasibility of PLA recycling remains less optimistic due to the lack of commercial capacity to cover the construction costs of PLA recycling plants. Nonetheless, research on PLA recycling remains an important part of the biodegradable polymer field, laying the groundwork for the establishment of future biodegradable polymer recycling technologies.

According to guidelines published by the American Plastics Recycling Association (APR), PLA has a Resin Identification Code (RIC) of 7 with the letters "PLA" to encourage the use of soft PLA in the mixed plastics recycling stream, as shown in Figure 9-1. APR is an organization that publishes detailed information on recycling different types of polymers in order to achieve high quality recycling of post-use materials. Typically, post-use PLA is divided into amorphous PLA and crystalline PLA. flake amorphous PLA is obtained from post-use items such as thermoformed, injection molded and blow molded parts. Also, semi-crystalline and crystalline PLA can be collected from oriented films or sheets or short or spunbonded fibers. when collecting PLA from post-use articles, it should be remembered that these articles should be of high quality before degradation (as it can be more easily degraded in a shorter period of time if it is biodegradable). This degradation can be determined by structural weakening, powdering, discoloration, fragmentation, or leakage. Degradation of the product after use is not recoverable and should not be mixed with the original PLA because it will harm the quality of the original PLA. For recovery, amorphous PLA and crystalline PLA must be dried at low temperatures of 43 to 55°C and 65 to 85°C, respectively. Two reasons why PLA needs to be dried prior to extrusion are: (i) to avoid PLA materials with low melting points from becoming sticky in the pre-extrusion dryer of the recycler, which may prevent PLA from flowing throughout the process; and (ii) to avoid the reaction between water molecules and PLA leading to hydrolytic degradation, which initiates the depolymerization process. In addition, mixing/blending of non-PLA materials with recycled PLA resins is strictly prohibited during the recycling process of PLA. Accidental mixing of PLA with other materials may occur due to the possible presence of non-PLA materials and coatings in the article after use. For example, PLA bottles may have printed polyethylene film labels attached to them, which need to be removed prior to the recycling process to avoid adverse effects on the polymer due to incompatibility of the polymer components. In addition, recyclers need to be aware that special additives can also adversely affect recycled polymers, such as antistatics, anti-stick agents, anti-fogging agents, anti-slip agents, UV stabilizers, impact modifiers, heat stabilizers, fillers and reinforcements. These additives need to be further tested to ensure compatibility when mixing different recycled resources.
9.7. Conclusion
PLA is a biodegradable polymer that can be blended into different grades of PLA resin using an extrusion process. The extruded PLA resin can be further processed into PLA part products through a variety of common processing methods such as blow molding, injection molding, thermoforming, and 3D printing. This chapter also discusses the processing conditions of PLA under various processing methods. The application of advanced technologies such as 3D printing and electron beam irradiation can improve the performance of PLA with complex shapes and designs. For recycling of PLA, collected post-use PLA items should be of good quality before degradation. Post-use PLA products that have degraded cannot be recovered by blending with virgin PLA resins, as this may adversely affect the product.
10. Applications of Polylactic Acid
10.1 Introduction
Polylactic acid (PLA) is a biodegradable polymer with a wide range of applications. Because of its biocompatibility and biodegradability in contact with mammals, it has decades of wide application in biomedical and pharmaceutical fields. However, due to the high cost of synthesis in the laboratory, the application of PLA has been very limited for many years. In most cases, PLA has been produced by direct polycondensation routes through lactic acid, resulting in PLA with low molecular weight and poor mechanical properties.
With the development of ring-opening polymerization production, the performance of PLA has been greatly improved. This pathway requires intermediate substances called propylenes. Propanediol is a cyclic dimer of lactic acid and can be a stereoconjugate in the form of L-propanediol, D,L-propanediol (racemic propanediol) and D-propanediol. Today, the synthesis of PLA rarely starts with the chemical synthesis of lactic acid. The lactic acid used is derived from the fermentation of carbohydrates such as starch and cellulose. A large portion comes from corn and cassava. Microbial-based fermentation produces mainly L-lactic acid.
Currently the largest producer of household PLA, NatureWorks produces 150,000 tons per year of PLA under the brand name Ingeo, using propylene glycol ester ring-opening polymerization technology. natureWorks' PLA is used in the production of biodegradable packaging, containers, clothing, fibers, etc. Total-Corbion is a leading producer of lactic acid, its derivatives and PLA for the biomedical and pharmaceutical industries.
This chapter summarizes the product applications of PLA, which can be divided into three main categories: household, pharmaceutical/biomedical, and 3D printing and engineering. Products, trade names and manufacturers are included in this document. The purpose is not to advertise, but to provide supporting information and references.
10.2 Household polylactic acid
Most of the PLA produced worldwide is used in household products such as clothing, bottles, cups and food packaging items. The goal of these PLA products is to replace existing petroleum-based chemical polymers, with the advantage that PLA products have an environmentally friendly production process and biodegradable waste. The applications and capabilities of PLA have evolved significantly. The authors believe that consumers are fully aware of the benefits of PLA and that PLA is no longer seen as a specialty polymer with limited applications.
Table 10-1 Household polylactic acid
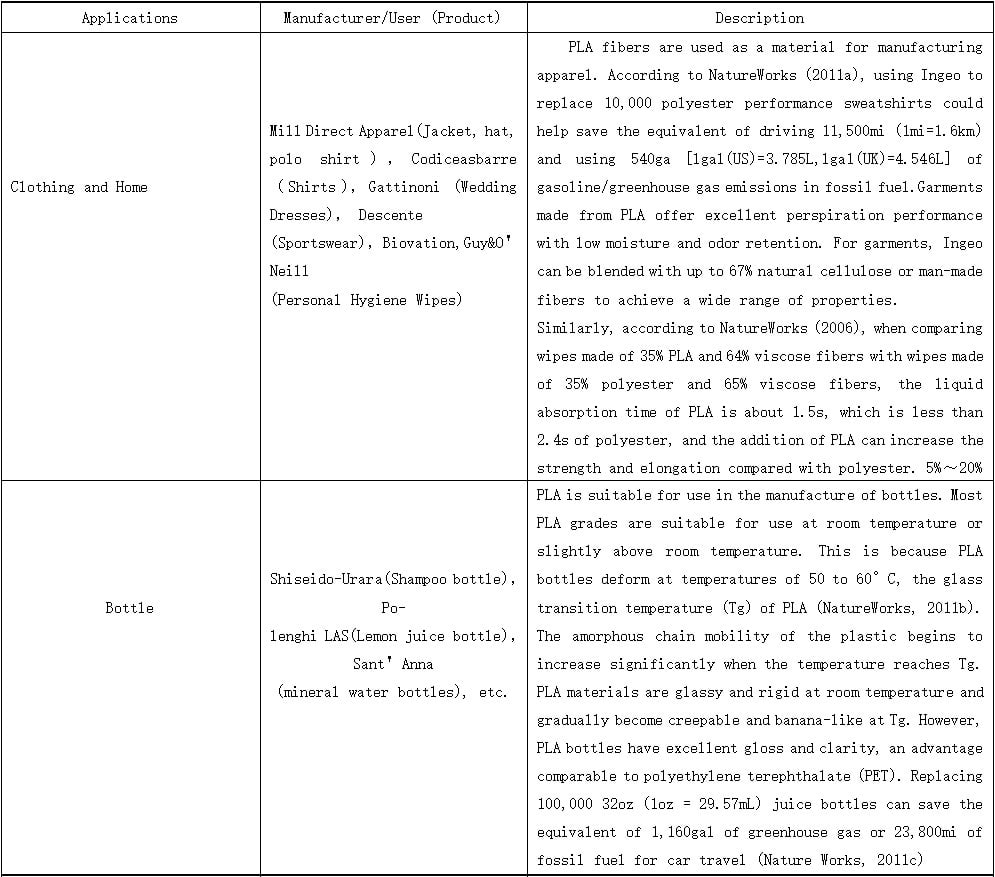
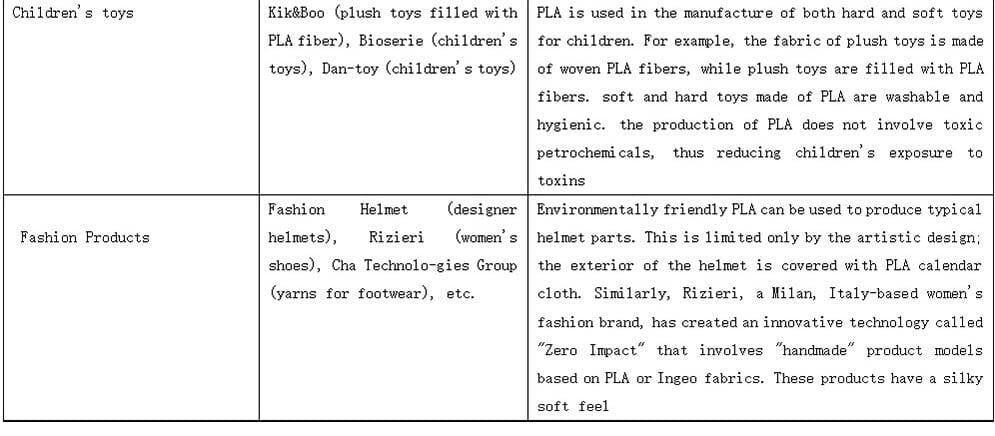
10.3 PLA Applications in 3D Printing, Engineering and Agriculture
The rigidity of PLA ensures good mechanical properties during use and easy biodegradation after disposal. The use of PLA in basic engineering parts is limited. As shown in Table 10-2, the use of PLA is primarily focused on auxiliary applications. For its use in electrical and electronic applications, Table 10-3 shows a comparison of PLA and PVA coated cables.
Table 10-2 PLA Applications in Engineering, Agriculture and Health
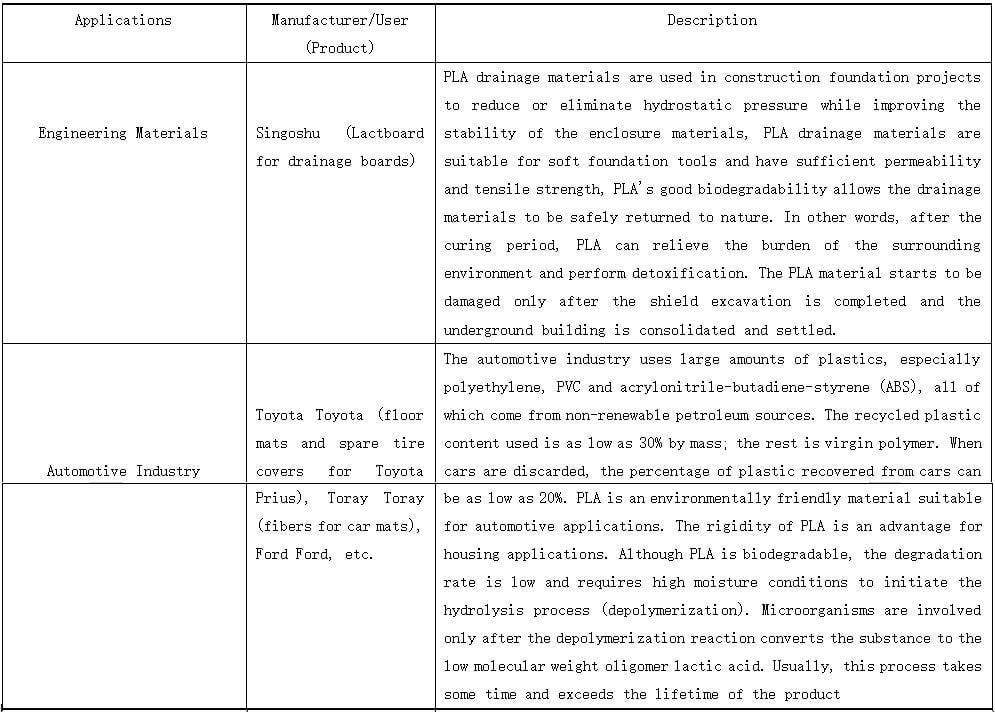

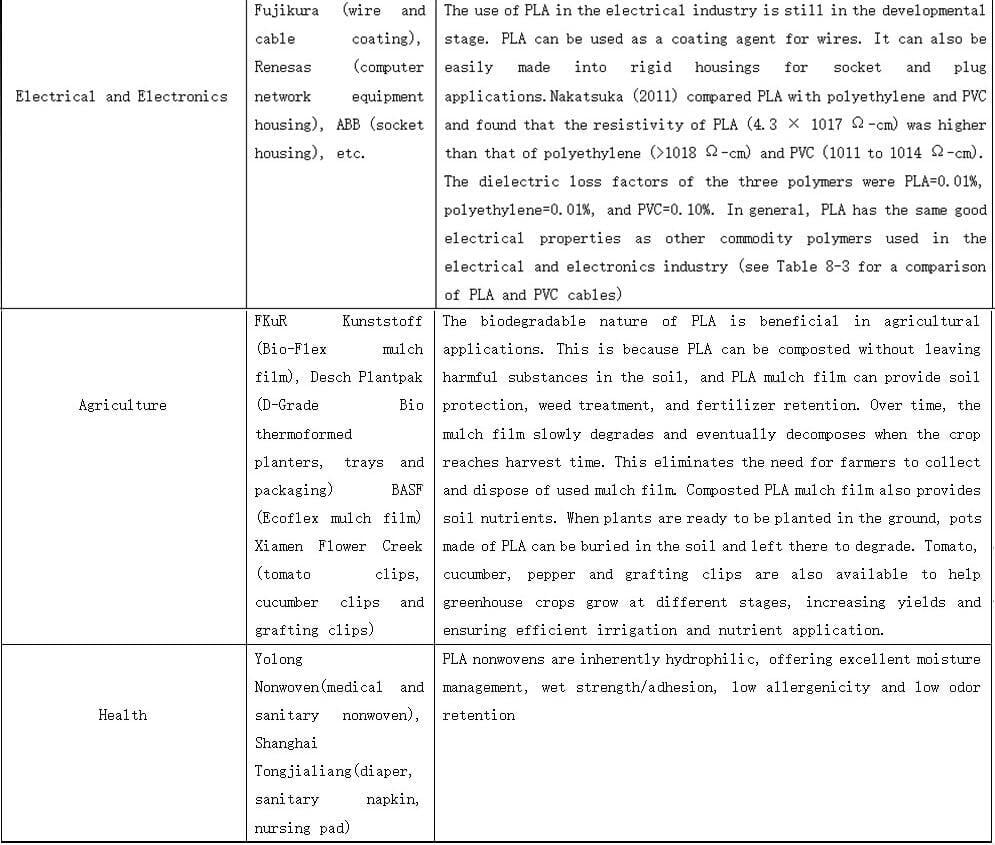
Table 10-3 Evaluation of PLA-coated cables compared to PVC-coated cables (Nakatsuka, 2011)

10.4 Polylactic Acid in Biomedical Applications
In the early stages of PLA development, most of its applications were in the biomedical field. PLA continues to be used in this field (Table 10-4). It is widely used in scaffolds to provide temporary structural support for the attachment and growth of tissue during surgery. It is also used as a drug carrier containing controlled-release active agents for long-term therapy, including cancer.
Table 10-4 Biomedical Applications of PLA
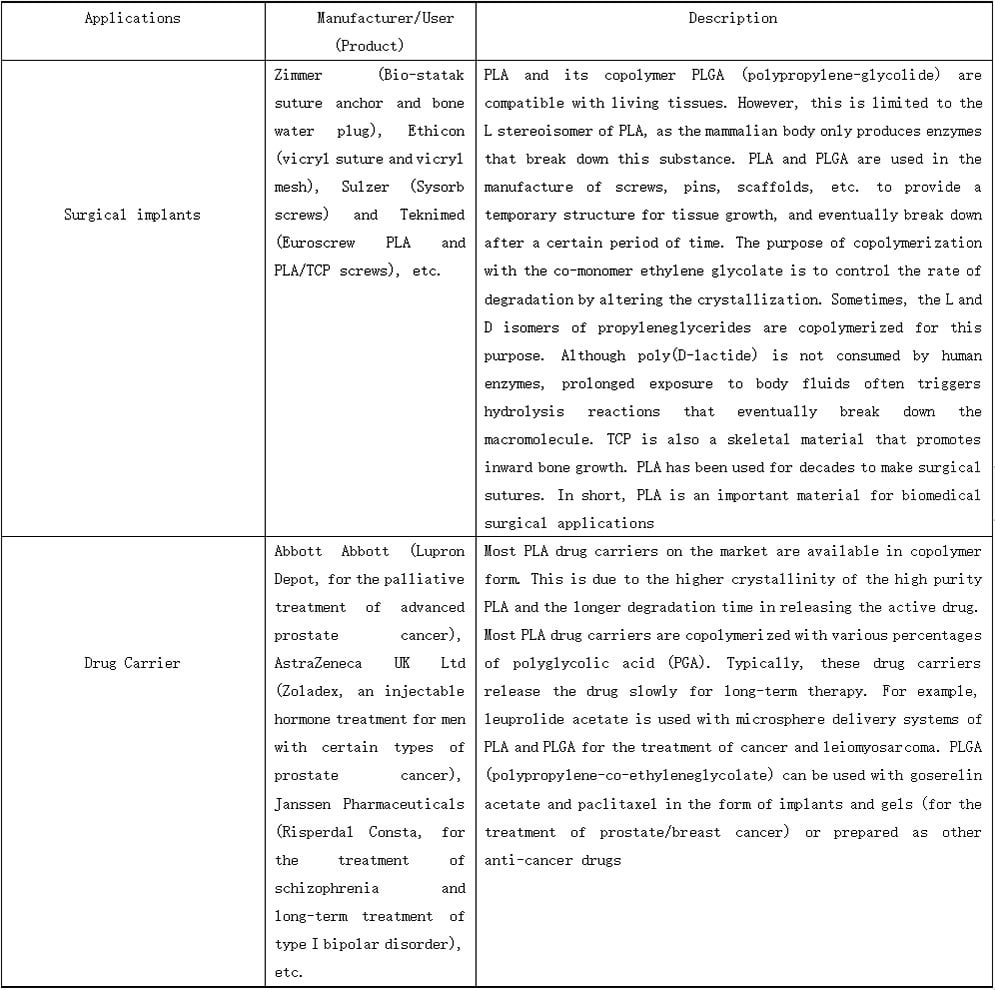
10.5 Conclusion
PLA is a very useful polymer that has found established use in many industries. PLA is biodegradable and environmentally friendly, making it well positioned in niche markets. Its use in biomedical and pharmaceutical applications dates back decades. The development of PLA applications in recent years has been primarily concerned with addressing environmental issues and reducing the negative impact of using non-biodegradable petrochemical polymers. The use of PLA has increased significantly in the consumer goods market in general, and in biodegradable packaging in particular. Significant developments are predicted for PLA, making PLA as economical as commodity plastics but with more environmentally friendly properties.
11.Environmental Assessment and International Standards for Polylactic Acid
11.1 Introduction
In recent decades, various types of biodegradable polymers have entered the consumer market, such as polycaprolactone, polyhydroxybutyrate, polyhydroxyvalerate cool, polylactic acid (PLA), and polyhydroxyalkanoate. Among commercially available biodegradable polymers, PLA is widely accepted by consumers due to its easy availability and flexibility, and is used to make a variety of polymer products to replace existing non-biodegradable petroleum-based plastic materials, namely polystyrene (PS), polyethylene, polypropylene, or polyvinyl chloride. Overall, life cycle assessment (LCA) is the most common tool used by researchers to assess and compare the ecological properties of PLA with other materials commonly used as disposables, such as aluminum, metals and paper. LCA is now widely used to assess the "cradle to factory" and "cradle to grave" carbon footprint (CF) of products, materials, processes and systems. This information is essential to determine what improvements can be made to minimize the environmental impact of a product. In other words, LCA is a meaningful analysis that thoroughly differentiates PLA applied to the manufacture of consumer products from other materials on the market, thereby protecting the environment.
11.2 Ecological profile of PLA and comparison with other materials
Over the years, researchers have conducted many LCA analyses, and the cumulative publication of PLA's LCA is shown in Figure 11-1. This information can be used to compare the environmental impact of various products. Because PLA is sourced from agriculture, it is often considered to be an environmentally friendly product. However, several factors can affect the environmental impact of plastic products, such as recycling, ability to reuse, cleanliness requirements for post-use items, transportation, and whether to dispose of them by incineration and landfill or something else at the end of their useful life.
In the study by Simon et al., they compared aluminum, polyethylene terephthalate (PET), PLA cases, and glass beverage bottles. Notably, the used bottles produced the least amount of greenhouse gases (GHG) through recycling methods after the first use, i.e. recycling the bottles into a second material included adding them to the original material to save costs. The difference in GHG emissions between recycling, incineration and landfills can be as high as 7.64 times, especially for aluminum bottles. Simon et al. reported that PLA has the lowest GHG emissions with 66 kg CO2 equivalent, followed by a large 1.5 liter PET bottle with 85 kg CO2 equivalent and a case with 88 kg CO2 equivalent. In this case, PLA seems to be the most environmentally friendly product. However, when PLA bottles are incinerated and landfilled, GHG emissions may increase several times, reaching 498 CO2-equivalent and 500 CO2-equivalent, respectively. This evidence strongly conveys the message that recycling is the ultimate way to keep the environment greener, while incineration and landfills should only be considered at the end of a material's life. Nevertheless, the dramatic increase in GHG from incineration and landfills is similar to that of other materials such as glass, PET, aluminum and cardboard boxes.
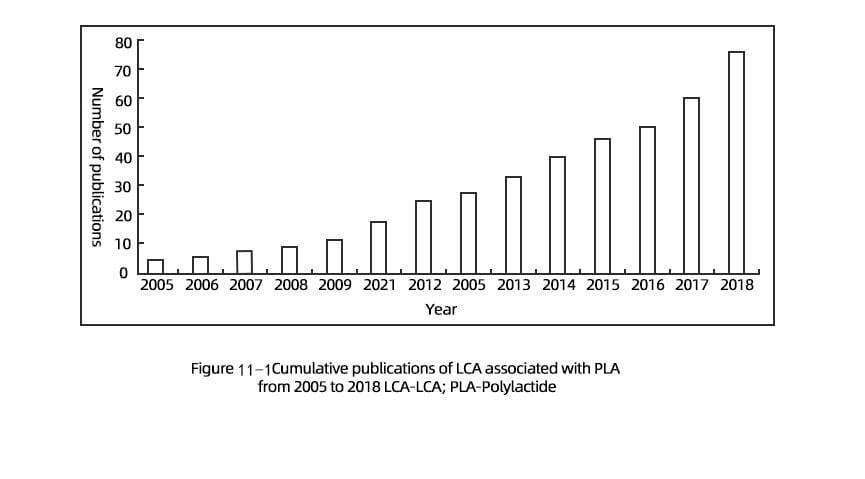
Another study conducted by Papong et al. compared PLA and PET drinking water bottles. They reported that the overall impact of PLA was lower compared to the production of PET bottles, but the eutrophication and acidification potential of PLA was higher. This is due to the fact that PLA is based on starch to produce the lactic acid intermediate monomer used for PLA polymerization. Fertilizers, herbicides and diesel fuel are needed to harvest and transport the starch produced by growing cassava roots from farms. Rainwater flushing fertilizers can pollute the water sources of lakes and rivers. In contrast, in the production of PET drinking water bottles, the emissions are limited to hydrocarbons, chemicals, catalysts and electricity as the main energy source for the production plant and there is no eutrophication. Nevertheless, PLA production has advantages in utilizing multiple sources of energy supply, such as burning agricultural residues to produce greener energy, and large areas of farmland where wind turbines can be applied to harvest wind energy and reduce reliance on non-renewable fossil fuels. Papong et al. summarized the production systems for PLA and PET in detail. As can be seen, the production process of PLA is very complex compared to PET, and requires the consideration of various other elements such as fertilizers, herbicides and enzymes as inputs to PLA, which in turn has similar inputs to PET such as: fuel, electricity, various chemicals, water and catalysts. This also explains the wider area involved in the production of PLA compared to PET, which may cause pollution.
Cherennet et al. studied in detail the water impact of producing bio-based boxes using different materials such as PS, PLA from sugarcane derivatives (PLA-S), starch-blended sugarcane derivatives PLA (PLA-S /starch) and polybutylene succinate (PBS). One of the most interesting parts of this analysis is the water footprint (WF) assessment, which is further divided into three types of WF: green WF, blue WF, and gray WF. green WF is the ratio of rainfall to field crop yield during the growing period, while blue WF is the ratio of irrigation water to field crop yield during the growing period. Meanwhile, the gray WF is the amount of water needed to dilute the pollutant concentration. This detailed analysis shows that PLA-S actually consumes more water to produce bio-based boxes, since growing sugarcane requires a lot of rain and irrigation water. As a result, the WF of PLA-S was 1.11 m3 , of which 36.14% was green water, 49.82% was blue water and 14.04% was gray water. PS as a non-renewable polymer material requires 0.70 m3 of water, 100% of which comes from blue water. This is followed by PLA-S/starch mixed with starch, which requires 0.55 m3 of water per box, and PBS, which has the smallest WF of 0.38 m3. Interestingly, the addition of starch to PLA-S was able to reduce the WF in PLA-S by 50.42%, mainly due to the higher amount of water required to produce sugarcane compared to cassava starch Cheroennet et al. further confirmed that the irrigation water used to grow sugarcane was significantly higher than cassava, which resulted in blue WF of 0.55 m3 and 0.27 m3 for PLA-S and PLA-S/starch, respectively. The gray water of LPA-S is also higher than PA-S/starch because PLA-S requires 4.06 kg per box, while sugarcane and cassava require 1.75 kg and 0.11 kg per box, respectively. this can be explained by the fact that the mixing of starch is a straightforward process, where the raw starch is added to PLA-S as a filler. However, the production of PLA-S involves a reaction in which multiple conversion stages may lead to significant mass loss. Therefore, PLA-S/starch is advantageous as far as the production of bio-based boxes with less material consumption is concerned. In addition, Cheroennet et al. found that between PS (0.05 kg equivalent CO2) and PLA-S-starch (0.303 kg equivalent CO2), PLA-S had the highest CF of 0.675 kg equivalent CO2. these data come from the observations of Cheroennet et al. that box formation is also highly dependent on the amount of plastic material used to manufacture the boxes. For example, because of the very low density of PS foam boxes (about 0.053 kg PS/box), PS boxes can be produced with very small amounts of resin. In comparison, PLA-S requires about 0.243 kg of PLA-S to produce PLA-S cartridges, followed by 0.105 kg of PLA-S pellets and 0.032 kg of tapioca starch to make PLA-S starch cartridges. It also shows that the environmental friendliness of plastic boxes depends not only on the choice of materials, but also on the following factors: (i) the amount of materials used, (ii) water resources, (iii) complexity of the production process, (iv) transportation of raw materials to the factory, (v) delivery distance to the consumer, and (vi) recyclability and reusability, which may affect the environmental footprint of plastic products. Transportation factors are the most important. For example, transporting PLA from a production site in Nebraska, USA, to Europe can generate significant amounts of fuel compared to local production, which can significantly reduce fuel consumption.
Overall, the issue of transportation needs to be thoroughly examined to demonstrate the environmental friendliness of plastic products, especially from the input of raw materials to the production of finished products.
More than a decade ago, NatureWorks, a major producer of PLA in the United States, revealed Life Cycle Analysis (LCA) based on corn. Initially, Vink et al. found in the first LCA that the main energy input required for PLA production was from fossil fuels, which was not attractive. To reduce the dependence on fossil fuels energy inputs should be improved and replaced by wind power and biomass. As shown in Figure 11-2, fossil energy consumption is 54.1 MJ/kg PLA, while renewable energy is 28.4 MJ/kg. This suggests that PI A production lacks justification in terms of renewable properties, despite the fact that the feedstock is derived from corn. Nevertheless, PLA is still superior to other petroleum-based polymers because it uses fewer petrochemical inputs, as shown in Figure 11-3. Vink et al. proposed an optimized solution for replacing fossil energy with biomass/wind energy that could further improve the environmental selling point of PLA.
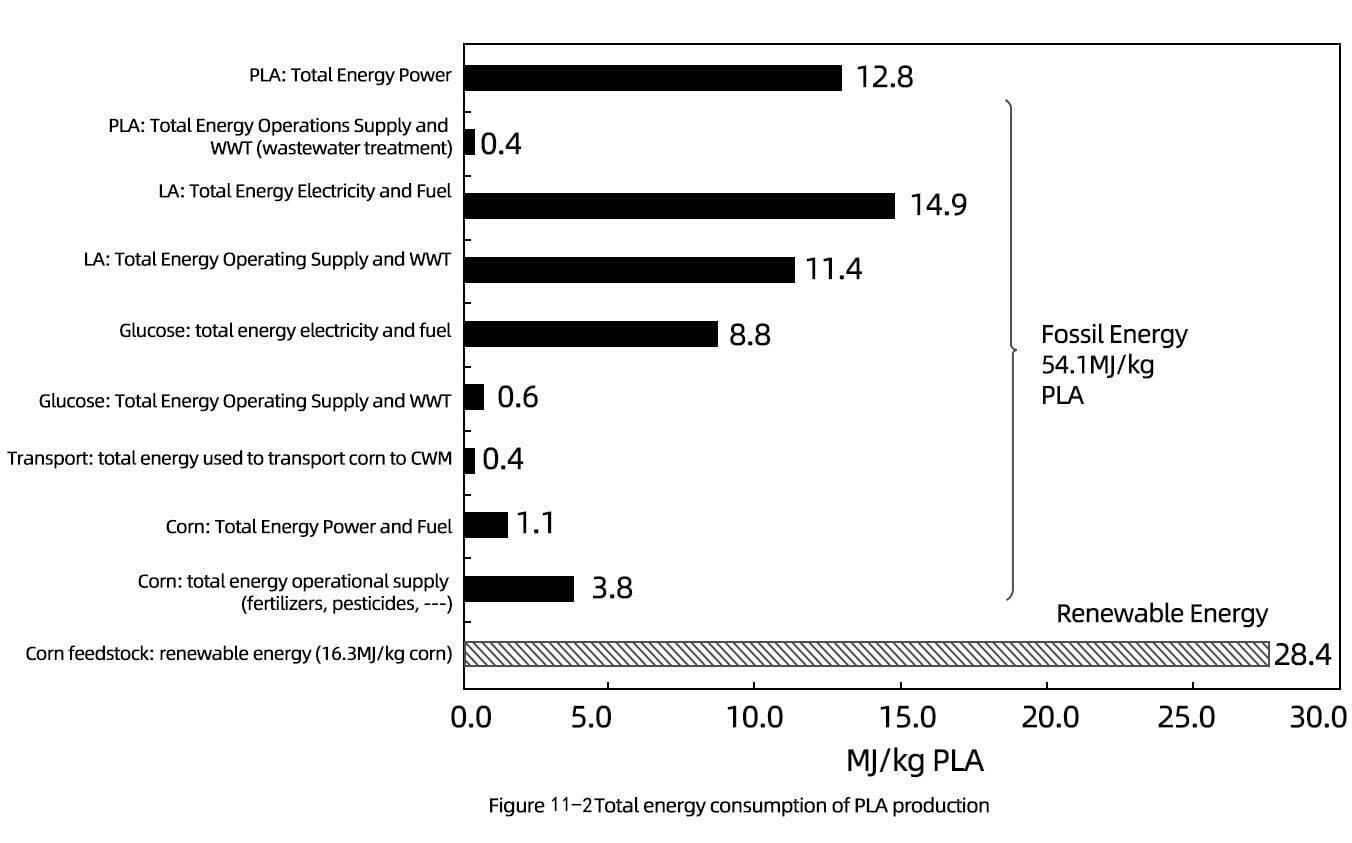
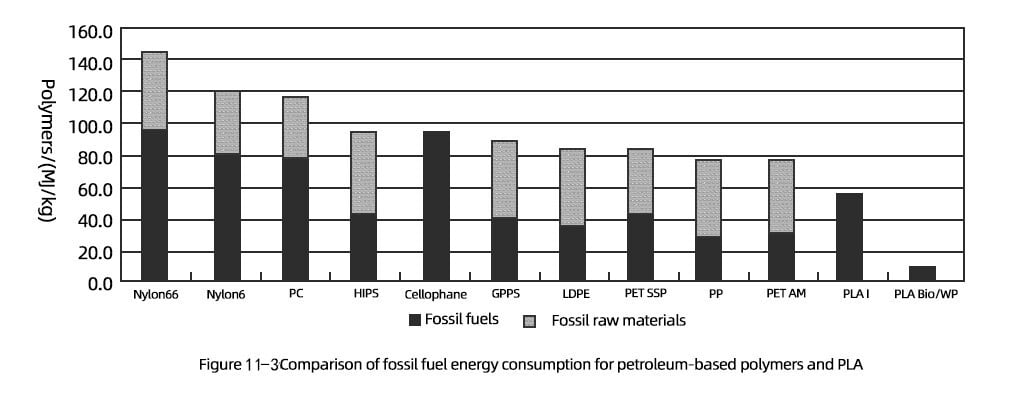
In subsequent PLA developments, NatureWorks has worked to reduce its reliance on fossil fuel inputs. natureWorks has spent years of research investigating the use of renewable energy sources. However, NatureWorks found that the PLA plant in Nebraska was not in a location where wind resources were economically competitive, and Nebraska is a public power state; in other words, NatureWorks had to purchase power from local obligations that did not come from green sources. To overcome this, NatureWorks chose Renewable Energy Certificates (RECs) to reduce (i) indirect emissions from power generation, 1.561 CO2 equivalent (kg/kg PLA), and (ii) indirect emissions from fuel, materials, corn production and recycling, 1.244 CO2 equivalent (kg/kg PLA). In effect, RECs are a type of carbon credit trading that encourages companies producing renewable energy to trade in a voluntary type of market, thereby promoting green energy. Carbon credits can be traded to industries that cannot efficiently produce RECs but are eager to participate in the renewable energy industry to promote lower emissions in their economic activities. As a result, Vink et al. reported a 90% reduction in carbon emissions after purchasing RECs. Vink et al. reported subsequent process improvements for NatureWorks.
In addition, the Pulak (currently known as Corbion) technology uses sugarcane as a feedstock for the production of lactic acid. Placid's lactic acid plant is located in Thailand and began operations in. In the early stages of development, most of the lactic acid was exported to Plac's propylene-based plant in Spain for conversion, and Groot and Borén reported 500 kg of CO2 emissions per ton of PLA in their LCA assessment of sugarcane produced propylene-based and PLA in Thailand.
Although alternative renewable energy sources can be obtained by burning bagasse in the range of 17 to 95 kWh per ton of sugarcane, Groot and Boren highlight that the variation in environmental credits depends on the type of by-product, the combustion technology and the energy mix in the application. In other words, each source of PLA has unique ecological characteristics and the selection of eco-friendly processes is critical to develop better green PLAs. Secondly, the most detrimental effect fraction of PLA still comes from the process of sugarcane cultivation and conversion to sugar. In addition, the cultivation of sugarcane contributes significantly to eutrophication, acidification and photochemical ozone production due to nitrogen emissions from amino fertilizers. During the combustion of agricultural residues used for cogeneration, greenhouse gases such as NOX, SOX and CO are released. Some activities associated with microbial production in the soil may also lead to NOX and methane emissions. the production of PLA is due to the impact of continuous replanting on agricultural land, causing soil erosion and loss of natural nutrients. Therefore, precautionary measures and environmental assessments are needed prior to deforestation for sugarcane planting.
PLA is a suitable alternative to existing petroleum-based polymers for the manufacture of cups, containers and packaging products. PLA is known to be degradable when disposed with municipal waste, thus reducing the environmental burden. Unlike petrochemical polymers such as PE, PP, PET, PC and PS, which take hundreds of years to decompose into harmless substances, PLA is fully compostable and widely accepted as a green product, especially in Japan, the United States and the European Union. Several reports have been published on the eco-efficiency of PLA post-consumer products compared to conventional plastics. These include cups (Vercalsteren et al.), flip-top packaging (Kruruger et al.), and wrapping paper (Hermann et al.).
An ecological analysis comparing the use of four plastic cups-reusable PC cups, disposable PP cups, disposable PE-coated paper cups, and disposable PLA cups-at public events in Flanders (Belgium) was reported by Vercalsteren et al. to the Flemish Institute of Technology (VITO), concluding that there is no clear indication of which cup system has the highest or lowest environmental impact. A direct comparison of the impact categories (carcinogens, ecotoxicity, fossil fuels, etc.) to show the superiority of the cup system over other cups is less convincing. For example, PLA cups use less fossil fuel than PP cups, but respiration caused by inorganic substances is still highest for paper cups with PE coating. In addition, the different scales of activity can affect the eco-efficiency of the cups. When used at small events, PC cups have minimal environmental impact. This is due to the fact that reusable PC cups can be hand-washed with only a small amount of water and detergent during the cleaning process. In contrast, PC cups have a higher turnover rate at large events and wear out more quickly during the subsequent frequent cleaning process, requiring regular replacement. Although PLA cups have the highest ecological specifications, as the current technology development for PLA is still in its infancy, certain environmental aspects (e.g. acidification/eutrophication and dependence on fossil fuels) need to be addressed in order for PLA to be competitive in the long term in applications. Such initiatives have yielded fruitful results in terms of the ecological improvements NatureWorks has made to the second-generation Ingeo (PLA6), which is 20% lower than the first-generation PLA cup (PLA5). NatureWorks aims to offer Ingeo (PLA/NG) as the absolute green product for better environmental protection.
The Institute for Energy and Environmental Research (IFEU) in Heidelberg, Germany, compared the head-to-head life cycle of flip-top packaging made from Ingeo, new PET, and recycled PET. A report by Kruger et al. compares the environmental loads when using landfill and incineration methods for the treatment of flip-top boxes. Both methods are commonly used in Europe and the United States. Eutrophication and acidification of water bodies appear to be higher in Ingeo, mainly due to the production phase involving agricultural and soil activities, which generate greenhouse gases. Although recycled PET appears to be a green product compared to PLA, recycled PET is actually derived from new material PET, where the upstream manufacturing process has been offset in the calculation of new material PET. Therefore, it is strongly believed that Ingeo can also yield better ecological performance in terms of recycling.
11.3 Environmental profile and global warming potential of polylactic acid food packaging
As mentioned earlier, the extent of the environmental benefits of PLA also depends on its application. Because PLA is the world's most produced biodegradable polymer and is reasonably priced, many food industries, especially those involved in single-use applications such as food packaging, use PLA as a food packaging material. ingrao et al. analyzed PLA foam trays using a 100-year global warming potential (GWP100) and found that the production of PLA foam trays typically require a total mass of 4.826 kg equivalent of CO2. PLA resin itself accounts for 61.26% of the CO2 generated, followed by transportation of PLA resin to the pallet manufacturing plant at 14.33%, and finally 11.63% from process electricity use. In comparison to polystyrene foam, Ingrao et al. (2015a) also found that GWP100 expandable PS was 5.11 kg equivalent CO2, a small difference from the 4.826 kg equivalent CO2 of PLA foam trays. This is because PLA is plant-derived and requires significant interdependence between agricultural and industrial activities to ensure high crop yields. In addition, the long distances between PLA production plants and final product processing facilities can further create additional environmental burdens due to transportation emissions.
In another study, Leejarpai et al. further explain that the environmental impact of PLA also depends on whether land changes are considered. Land use change is a factor to be considered when converting land to crops, which may involve a large number of processes such as decomposition, nitrification/denitrification, photosynthesis and combustion, all of which contribute to global warming. Figure 11-4 shows that land use change PLA ("PLA") has the highest CO2 emissions, followed by PET. nevertheless, Leejakpai et al. reported that PLA biodegrades better than PS under landfill conditions.
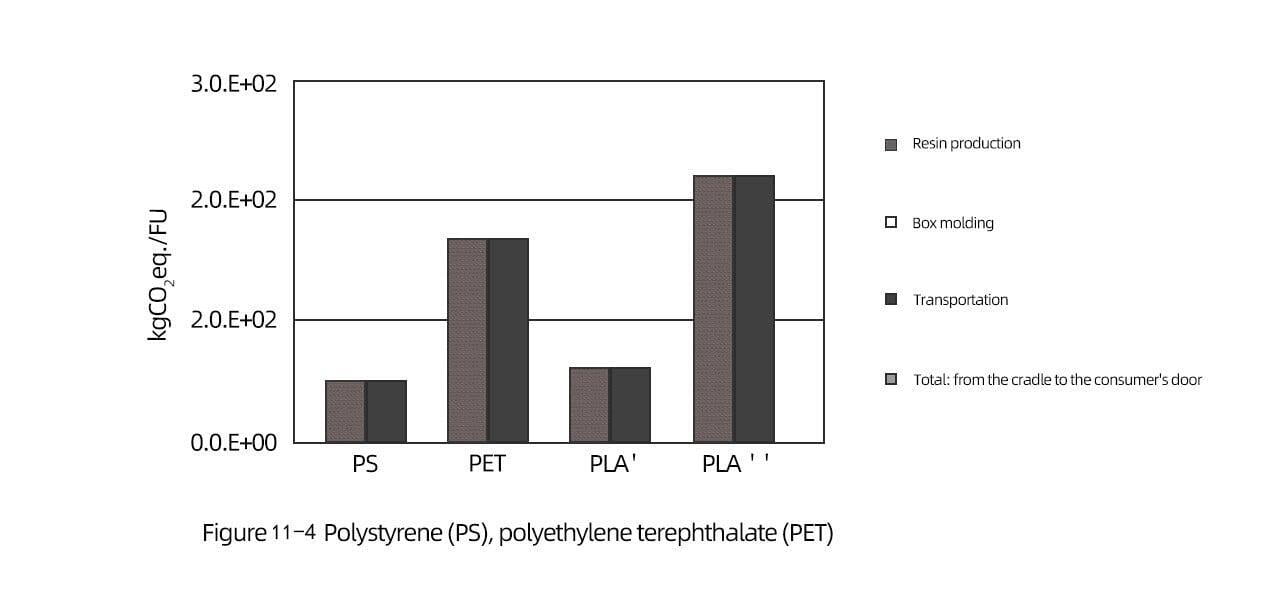
PLA sheets underwent complete biodegradation and structural destruction after 6 months of burial, while PS did not show any significant difference even after 20 months of burial. This biodegradable property is advantageous in reducing plastic pollution caused by non-degradable plastics.
11.4 List of International Standards for Biodegradable Polymers
Several major standardization organizations [International Organization for Standardization (ISO), American Society for Testing and Materials (ASTM), European Committee for Standardization (CEN) and German Institute for Standardization (DIN)] have developed several standards.
11.5 Conclusion
PLA is a biodegradable polymer that offers great advantages in overcoming waste plastic pollution. In addition, the inputs used to produce PLA come from agriculture, giving it additional advantages as a renewable polymer compared to petroleum-based polymers. On the other hand, the environmental friendliness of PLA products remains a controversial issue, as agricultural activities can also lead to water pollution, the use of electricity that consumes fossil fuels, and the use of fertilizers, herbicides/pesticides, etc. that can lead to other types of pollution. More importantly, the transportation of PLA pellets from the plant to the processing site can further contribute to carbon emissions. Therefore, taking all these factors into account, the challenge of reducing the carbon footprint of PLA remains enormous. Researchers need to be urged to continuously study to reduce the environmental impact of PLA, especially during the production process and the processing of the final product. This will increase the feasibility of replacing petroleum-based polymers with PLA in the future.
